Hi,
I am trying to simulate an ammonia water mixture. Below are the methods I am trying to use:
-
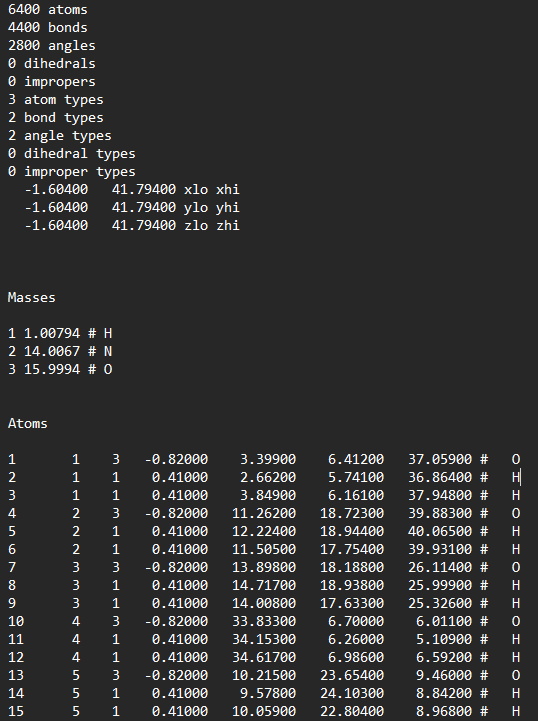
Generate an amorphous cell of ammonia and water with pcff forcefield in Materials Studio. Then exported it with .car and .mdf files. Lastly convert the .mdf file into LAMMPS data file. But this method seems to unable to convert H-N-H-H bond so it became impropers.
-
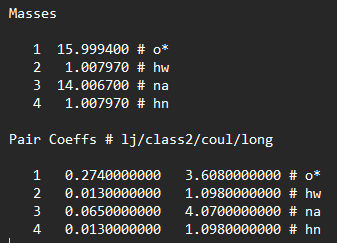
Generate an amorphous cell of ammonia and water without forcefield in Avogrado. Then use read_data command to add the data. But the system need the pair-coeff defined.
So, I would like to ask the experts here, any idea how to type the pair-coefficients? As you can from both screenshots, the masses are defined differently. Or anyone experts can teach me how to correct the impropers?
Thank you.
There are quite a few external builder tools available that are compatible with LAMMPS for a selection, please see: https://www.lammps.org/prepost.html
It is also possible to construct systems from using molecule files and the create_atoms command — LAMMPS documentation
I the few cases that I have simulated ammonia in water, I had started with a system of pure water and then wrote a little program that would modify the corresponding data file to replace some of the water molecules with ammonia (and a corresponding number with an anion like Chloride, so the system remains neutral).
There is not much else that I can advise, since the remainder of your problems are not really LAMMPS problems, but problems of understanding and researching the physics of your model.
The syntax of how you enter masses and potential parameters is described in the manual. What are the suitable parameters for your specific problem is a different story. That you can learn from researching the published literature.
I the few cases that I have simulated ammonia in water, I had started with a system of pure water and then wrote a little program that would modify the corresponding data file to replace some of the water molecules with ammonia (and a corresponding number with an anion like Chloride, so the system remains neutral).
This is an interesting idea. I have successfully run the script but the result is far from what I imagine. Since I can’t upload file due to being new user, simply said, the result shown in VMD is just zooming in to my simulation box.
Will you be kind to teach me how you do it? Or is it because I’m reading the whole amorphous cell created from Materials Studio into LAMMPS causing it to see the whole amorphous cell as 1 atom?
Thank you.
if I understand your problem correctly, the coordinates were not processed correctly, and as a result, you only see one atom in VMD. You can’t upload files, but you can add a snipped of code showing the structure of your data file, eg
512 atoms
512 ellipsoids
1 atom types
-12.437 12.437 xlo xhi
-12.437 12.437 ylo yhi
-12.437 12.437 zlo zhi
Atoms
1 1 11.07611 -2.15772 -1.07319 1 1 1.06190466
2 1 -4.53403 4.38844 -2.68447 2 1 1.06190466
3 1 10.77808 -0.34289 -5.84963 3 1 1.06190466
4 1 -4.98151 7.09881 5.67341 4 1 1.06190466
5 1 8.22049 2.41727 -4.91064 5 1 1.06190466
6 1 12.06951 8.92151 -7.89213 6 1 1.06190466
7 1 0.57682 4.86851 -11.79457 7 1 1.06190466
8 1 -9.24918 -5.88359 -0.93958 8 1 1.06190466
9 1 -0.20982 9.56309 6.33637 9 1 1.06190466
This forum uses markdown, plus it has a friendly GUI. Please make an effort to use it properly.
Sorry, but no. This is not the place to be taught how to write a program to parse and process data files. Either you have sufficient programming skills to do it or you don’t. There are pieces of code available that can read or write certain file formats that you can use and there is detailed documentation of relevant formats in LAMMPS. That should be more than sufficient to do this kind of processing.
What you mean is this right?
Seems like I have been promoted to trusted user so I can upload the file now.
input.lammps (1.2 KB)
log.lammps (90.8 KB)
Ammonia water mixture file:
mixture.lmpdat (625.3 KB)