Dear lammps user,
First my lammps version is 7 August 2019.
Second, I would like to randomly generate two-dimensional spheres inside a two-dimensional box with periodic boundary conditions and with a packing fraction 70 %. I wrote the below lammps script to do so.
In order to have a packing fraction of 70%, I am trying to increase the number of circles inside the fixed-area simulation box without getting the circles overlapped. The simulation box area generated by the below script is (141.42*141.42). In order to obtain 70% for such area, the number of circles should be at least 4432 approximately. However, the following script just generate 1505 circles and this gives packing fraction much less than 70%.
I set the delete_atoms cutoff distance to be 2 because any values less than 2 will get the circles overlapped.
Could you please tell me why I cannot add more particles to the 2-d box in order to have a packing fraction of 50% at least ?.
I am sorry if my question is simple, but I really tried to understand it. Maybe the problem arises form the choice of the random number set in the create_atoms command.
Please, there were trivial mistakes in th eprevious email and I modified it below.
Dear lammps user,
First my lammps version is 7 August 2019.
Second, I would like to randomly generate two-dimensional spheres inside a two-dimensional box with periodic boundary conditions and with a packing fraction 70 %. I wrote the below lammps script to do so.
In order to have a packing fraction of 70%, I am trying to increase the number of circles inside the fixed-area simulation box without getting the circles overlapped. The simulation box area generated by the below script is (141.42*141.42). In order to obtain 70% for such area, the number of circles should be at least 4432 approximately. However, the following script just generate 1505 circles and this gives packing fraction much less than 70%.
I set the delete_atoms cutoff distance to be 2 because any values less than 2 will get the circles overlapped.
Could you please tell me why I cannot add more particles to the 2-d box in order to have a packing fraction of 70% ?.
I am sorry if my question is simple, but I really tried to understand it. Maybe the problem arises form the choice of the random number set in the create_atoms command.
There are some questionable things about your input. For starters, the z-dimension is very large for a 2d simulation.
Secondly, if your circles have a diameter of 1, wouldn’t the center-to-center distance at which there is overlap between them be equal to 1 rather than 2?
Thirdly, your input script only generates 4432 atoms to begin with. How can you delete atoms from a collection of 4432 atoms and then expect to arrive at 4432 atoms again? You need to add some command that increases the number of particles again, or come up with a different way to generate your initial input.
Keep in mind that LAMMPS is very low-level in the sense that it will do exactly what you tell it to do and nothing more (or less), so I would recommend making sure that you understand what each step in your input does, perhaps by writing data files after every command, until you know for sure you are telling LAMMPS what you want to tell it.
Dear Stefan,
Thank for your reply. I had been thinking of the things that you questioned about before I asked my questions.
Following what you suggested, I have modified the script as it is shown below, and I attached a snapshot photo of the time-step 1000 which is the last time-step in the simulation. The number of circles generated in the photo is 5751.
However, the circles still overlaps. !!!
Could you please tell where the problem is ?.
My aim is to generate non-overlapping circles with packing fraction 70%, and my idea to achieve it is the following script using the pair_style zero where there is no interaction between circles.
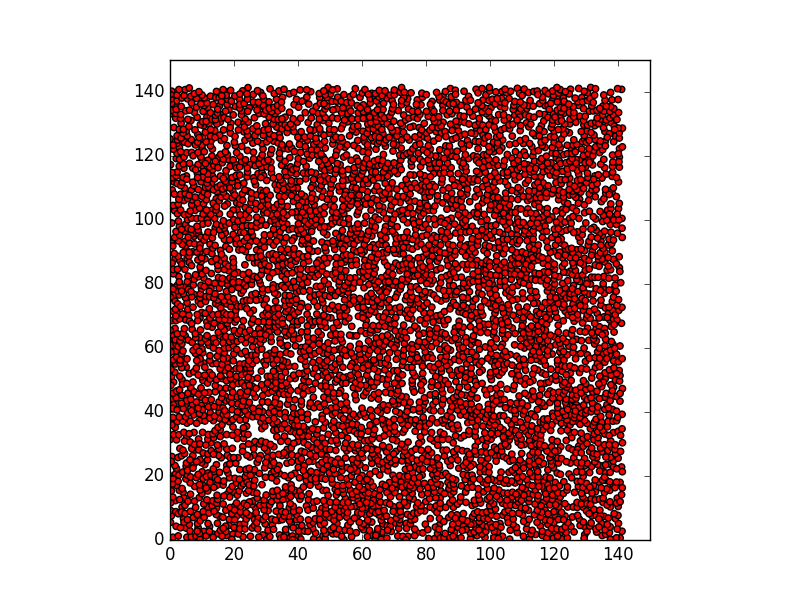
using pair style zero makes no sense. if particles have no interactions, they can move “through” each other without a problem.
as stefan was pointing out already, LAMMPS will do literally what you ask it to do, it doesn’t care about real life physics, only the rules that are programmed.
for the same reason, if you have, say, a Lennard-Jones interaction, the diameter of the particles will be completely ignored, since that property never enters anywhere into the potential energy function.
now for some productive suggestions. the easiest and most reliable way to get a system of a given density, is to generate a regular lattice. then you can compute ahead of time what the spacing between particles has to be. if you want to make it less regular, you can then “shake it up” by assigning (large) random velocities (if need be multiple times) until you have something reasonably random and mixed. of course, you will only avoid overlaps between extended particles, if you use a pair style, that takes the diameter/radius into account, e.g. from the GRANULAR package. please also keep in mind, that LAMMPS’s architecture, does not - in its current form - support hard sphere (or hard disk) models.
axel.