Hi axel
I have used vmd to get the whole molecule with following processes.
(1)get the dump from lammps with this command
dump 2 all custom 100 coro-x.lammpstrj id type q x y z ix iy iz
(2) directly load the coro-x.lammpstrj file to vmd
(3)type the following command in TK Console to get the whole molecule
pbc wrap –compound res -all
pbc box
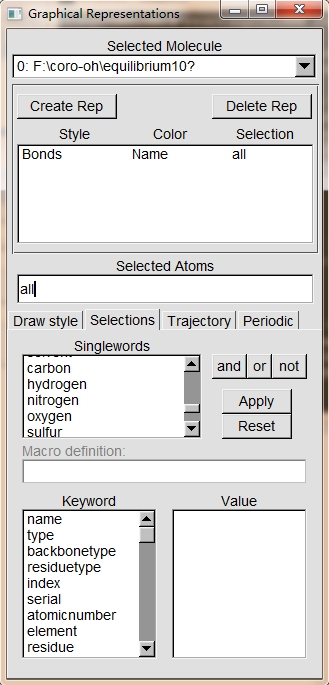
But I have another problem when I want to select hydrogen atoms with the
fllowing tab.
All atoms disappear when I choose "hydrogen" in Singlewords and click Apply
buttom.
My question is what should I do in Lammps, and then I can selet spicified
atoms in vmd with Graphical Representation?
you have to communicate to VMD that the atoms in question are hydrogen
atoms. your dump custom command identifies atoms with "type", which is
an arbitrary number with no particular meaning, so VMD doesn't know
what element that atom is. you can add "element" to the output list
for your custom style dump and use dump_modify element to define the
which type maps to which element.
This questions are also aked by me
http://sourceforge.net/p/lammps/mailman/message/34758681/.
i don't see a connection between this question and that one.
You said " in order to simply track the changes in the bond topology,
however, this is not needed, but rather a visual representation that can
recompute the connectivity would be sufficient. most viz programs compute
connectivity based on (rather simple) heuristics anyway, and for
visualization purposes it is rarely needed to follow the details as
meticulously."
I can recompute the connectivity based on cut-off distance in OVITO, but I
do not know how to recompute the connectivity based on "heuristics anyway".
the heuristics that VMD employs are rather simple: it is also cutoff
based, but it uses an element specific radius to determine the bond
cutoff. and for elements identified as hydrogen, only one bond is
allowed. this computation is automatically triggered when loading a
molecule and can be manually requested from the command line (mol
bondsrecalc <mol id>, see the VMD docs for details). there also is the
DynamicBonds representation, which employs a single global cutoff to
determine bonds on the fly. again, please read the VMD documentation.
Would you mind telling me how to do it in vmd?
this is really a question for the VMD mailing list. i've given you the
basic directions and told you where to look. any questions about
further details should be directed to the VMD mailing list.
axel.