I am now attempting to build the pivot algorithm for running Monte-Carlo simulations of polymers while simultaneously doing MD simulations. Crazy, I know.
My problem is:
Using PyLammps’ L.atom[i].position method, I have difficulty obtaining the unwrapped positions of each molecule/atom, i, after I have initialized the system with L.run(0).
Do you know how to obtain the unwrapped coordinates for each atom using PyLammps after the system has been initialized?
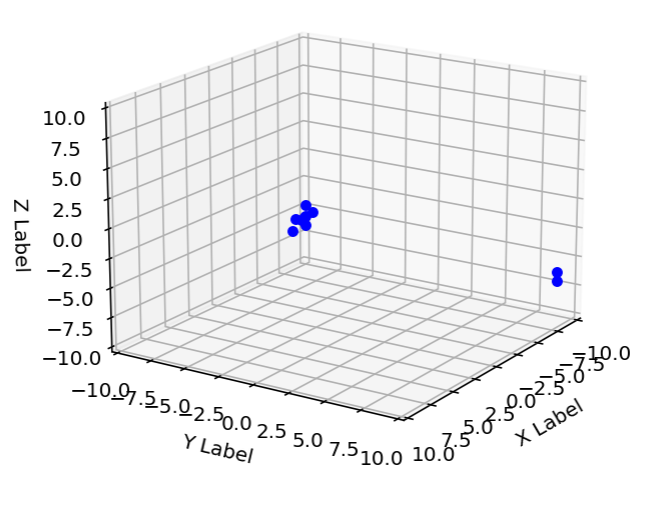
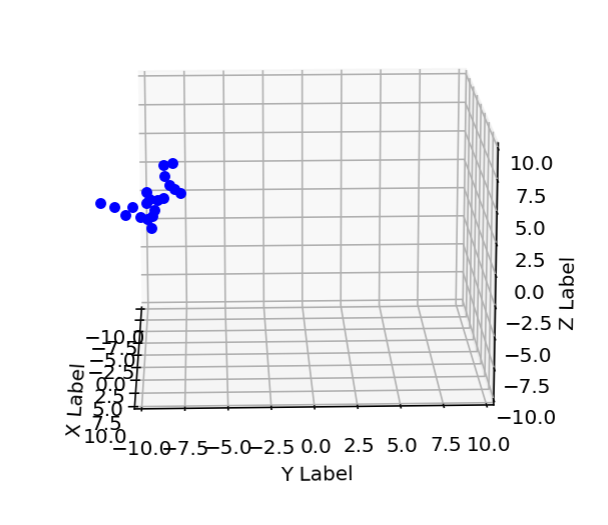
An example of a none self-avoiding polymer can be seen in the pictures I have produced:
First off, for any serious project, you should not be using PyLammps, but the regular LAMMPS python module. Due to its approach, PyLammps is very fragile and can be much slower.
Thank you for your help. You are entirely correct to not just using PyLammps.
As seen below, one can call a PyLammps Lammps instance.
It took me several hours to figure out.
Obtaining a lammps instance was a prerequisite for obtaining the unwrapped coordinates.
L = PyLammps() #PyLammps instance
LL = L.lmp #lammps instance
Then i could setup the simulation with PyLammps
L.compute("ucoordinates", "all", "property/atom", "id", "xu", "yu", "zu")
L.dimension(3) # 3D simulation
L.units("lj") # set units to Lennard-Jones
L.atom_style("molecular")
┊
L.create_atoms(1, "random", NPolymers*Nmonomers, 12345, "box") # create NPolymers*Nmonomers atoms in the box region with the given seed
┊
unwrappedxyz = LL.numpy.extract_compute("ucoordinates", LMP_STYLE_ATOM, LMP_TYPE_ARRAY)
To get it work, I had to utilize the lammps instance, L.lmp.
Also, strange behavior occurs when transferring atoms or molecules from one coordinate to another using PyLammps. When executing the code
for i in range(L.system.natoms):
L.atoms[i].position = (i*0.1 + 15, 15, 15)
L.run(1)
listucord = LL.numpy.extract_compute("ucoordinates", LMP_STYLE_ATOM, LMP_TYPE_ARRAY)
print(listucord[0][1], listucord[0][2], listucord[0][3])
L.run(0)
for i in range(5):
print("position of atom", i, "is", L.atoms[i].position)
print("unwrapped position of atom", i, "is", listucord[i][1], listucord[i][2], listucord[i][3])
for i in range(L.system.natoms):
L.atoms[i].position = (i*0.1 + 0, 0, 0)
L.run(1)
listucord = LL.extract_compute("ucoordinates", LMP_STYLE_ATOM, LMP_TYPE_ARRAY)
print(listucord[0][1], listucord[0][2], listucord[0][3])
L.run(1)
for i in range(5):
print("new position of atom", i, "is", L.atoms[i].position)
print("new unwrapped position of atom", i, "is", listucord[i][1], listucord[i][2], listucord[i][3])
Where the box is defined as
L.boundary("p p p") # periodic boundary conditions in all directions
L.region("box", "block", -boxSize/2, boxSize/2, -boxSize/2, boxSize/2, -boxSize/2, boxSize/2) # create a box region with the given box size
I get the output:
position of atom 0 is [-5. -5. -5.]
unwrapped position of atom 0 is 15.0 15.0 15.0
position of atom 1 is [-4.9 -5. -5. ]
unwrapped position of atom 1 is 15.1 15.0 15.0
position of atom 2 is [-4.8 -5. -5. ]
unwrapped position of atom 2 is 15.2 15.0 15.0
position of atom 3 is [-4.7 -5. -5. ]
unwrapped position of atom 3 is 15.3 15.0 15.0
position of atom 4 is [-4.6 -5. -5. ]
unwrapped position of atom 4 is 15.4 15.0 15.0
new position of atom 0 is [0. 0. 0.]
new unwrapped position of atom 0 is 20.0 20.0 20.0
new position of atom 1 is [0.1 0. 0. ]
new unwrapped position of atom 1 is 20.1 20.0 20.0
new position of atom 2 is [0.2 0. 0. ]
new unwrapped position of atom 2 is 20.2 20.0 20.0
new position of atom 3 is [0.3 0. 0. ]
new unwrapped position of atom 3 is 20.3 20.0 20.0
new position of atom 4 is [0.4 0. 0. ]
new unwrapped position of atom 4 is 20.4 20.0 20.0
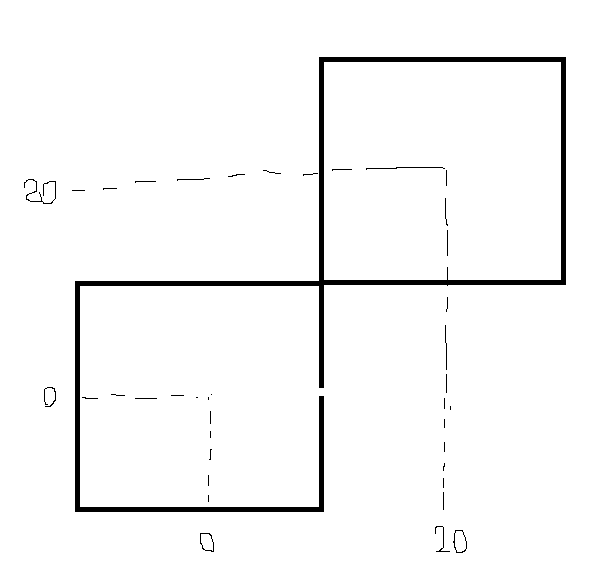
Therefore, while executing L.atoms[i].position = (i*0.1 + 0, 0, 0), the wrapped coordinates move near to the center of the original box (0*i,0,0), and the unwrapped coordinates travel close to the center of another box image (I am uncertain of the terminology) (20*i, 20, 20).
You are missing my point entirely. I am not suggesting to use the LAMMPS module in addition to PyLammps, I am recommending against using PyLammps altogether.
The inconsistency you see is likely part of that dual use.