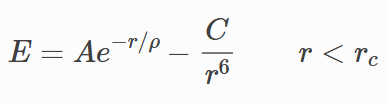Hi everybody.
I am trying to obtain amorphous silica structure at 2.2 g/cm^3. I am using BKS potental (LAMMPS version is 2020 March 3). I use two types of simulation.
- I use “read_data” to read the input quartz cell of 9 atoms and replicate it, and then after minimization I use “create velocity” command and then start melting and quenching with NPT. But my quartz instantly collapses then the density becomes even higher.
- I create atoms randomly inside a box (I tried different box dimensions to have initial density of 2.2, then smaller, then bigger) and relax them at room temperature. The density always gets bigger in the end then the amorphous density of 2.2 as expected. Plus, during the heating phase the density increases when I increase the temperature while it should be the opposite.
What could be the problem in both cases?
The script of first simulation:
variable num_dump_frames equal 100
variable num_particles equal 1944
variable temperature equal 300 # Initial temperature (K in metal units)
variable equilibration_steps equal 100000
variable pressure equal 1 # The pressure
variable cut_off equal 10 # Cut-off distance for the Buckingham term
units metal
atom_style charge
timestep 1e-3
read_data quartzNew.lmp
replicate 6 6 6
variable lhalf equal ((33.26*${num_particles}/1.5)^(1/3))/2
region orthobox block -${lhalf} ${lhalf} -${lhalf} ${lhalf} -${lhalf} ${lhalf} units box
variable num_silicon equal ${num_particles}/3
variable num_oxygen equal ${num_particles}/3*2
mass 1 28.085500
mass 2 15.999400
set type 1 charge +2.4
set type 2 charge -1.2
pair_style hybrid/overlay buck/coul/long ${cut_off} table linear 39901
pair_coeff 1 1 buck/coul/long 0.0 1.0 0.0 #No interactions between Si atoms
pair_coeff 1 2 buck/coul/long 18003.757200 0.205205 133.538100
pair_coeff 2 2 buck/coul/long 1388.773000 0.362319 175.000000 #BKS interaction in PRL 64 1955 (1990)
pair_modify shift yes
pair_coeff 1 2 table potential_SiO2.TPF Si-O ${cut_off}
pair_coeff 2 2 table potential_SiO2.TPF O-O ${cut_off} #See the potential file for more information
kspace_style pppm 1.0e-4
neighbor 3.0 bin
neigh_modify check yes every 1 delay 0 page 100000 one 10000
group Si type 1
group O type 2
thermo 1000
thermo_style custom step temp press pe vol fnorm density
thermo_modify lost warn
minimize 0 1.0e-8 100000 10000000
fix f0 all box/relax aniso 1.0 vmax 0.001
minimize 0 1.0e-8 100000 10000000
unfix f0
minimize 0 1.0e-8 100000 10000000
velocity all create ${temperature} 123
dump 1 all custom 1000 everything.dump id type x y z
fix f1 all npt temp ${temperature} ${temperature} 1 iso ${pressure} ${pressure} 1
restart 100000 TTM_pre_restart_*.mpiio
run ${equilibration_steps}
unfix f1
And here is the output:
LAMMPS (3 Mar 2020)
Reading data file ...
triclinic box = (0 0 0) to (4.916 4.25738 5.4054) with tilt (-0.458 0 0)
4 by 4 by 6 MPI processor grid
reading atoms ...
9 atoms
read_data CPU = 0.0285988 secs
Replicating atoms ...
triclinic box = (0 0 0) to (29.496 25.5443 32.4324) with tilt (-2.748 0 0)
4 by 4 by 6 MPI processor grid
1944 atoms
replicate CPU = 0.00780056 secs
Setting atom values ...
648 settings made for charge
Setting atom values ...
1296 settings made for charge
WARNING: 1 of 39901 force values in table are inconsistent with -dE/dr.
Should only be flagged at inflection points (../pair_table.cpp:483)
648 atoms in group Si
1296 atoms in group O
Setting up cg style minimization ...
Unit style : metal
Current step : 0
Per MPI rank memory allocation (min/avg/max) = 5.423 | 5.428 | 5.438 Mbytes
Step Temp Press PotEng Volume Fnorm Density
0 0 -18048.914 -35197.989 24436.329 290.18526 2.6457518
66 0 -118610.69 -36922.576 24436.329 0.15372121 2.6457518
Loop time of 0.303545 on 96 procs for 66 steps with 1944 atoms
Setting up cg style minimization ...
Unit style : metal
Current step : 66
WARNING: Energy due to 3 extra global DOFs will be included in minimizer energies
Per MPI rank memory allocation (min/avg/max) = 5.423 | 5.428 | 5.439 Mbytes
Step Temp Press PotEng Volume Fnorm Density
66 0 -118610.69 -36922.576 24436.329 0.15372121 2.6457518
128 0 -18.874279 -37041.512 23204.204 48.317312 2.7862392
Loop time of 0.269849 on 96 procs for 62 steps with 1944 atoms
Setting up cg style minimization ...
Unit style : metal
Current step : 128
Per MPI rank memory allocation (min/avg/max) = 5.425 | 5.43 | 5.439 Mbytes
Step Temp Press PotEng Volume Fnorm Density
128 0 -14.441756 -37041.499 23204.204 48.317253 2.7862392
145 0 -58585.793 -37076.291 23204.204 0.34455231 2.7862392
Loop time of 0.15236 on 96 procs for 17 steps with 1944 atoms
Setting up Verlet run ...
Unit style : metal
Current step : 0
Time step : 0.001
Per MPI rank memory allocation (min/avg/max) = 5.567 | 5.572 | 5.582 Mbytes
Step Temp Press PotEng Volume Fnorm Density
0 300 -55117.537 -37076.291 23204.204 0.34455231 2.7862392
1000 665.54568 -2714.7088 -37203.03 21876.836 83.854833 2.9552931
2000 640.07549 6008.503 -37263.761 22401.288 97.913217 2.8861047
...
98000 294.91862 1273.3414 -37408.209 22885.312 74.316524 2.8250637
99000 311.57424 1282.3418 -37410.219 22921.862 72.48088 2.820559
100000 305.01153 -231.2718 -37406.47 23012.551 73.820079 2.8094436
Loop time of 197.762 on 96 procs for 100000 steps with 1944 atoms
Performance: 43.689 ns/day, 0.549 hours/ns, 505.658 timesteps/s
Total wall time: 0:03:19
Here are the snapshots at 0 ps and at 1 ps
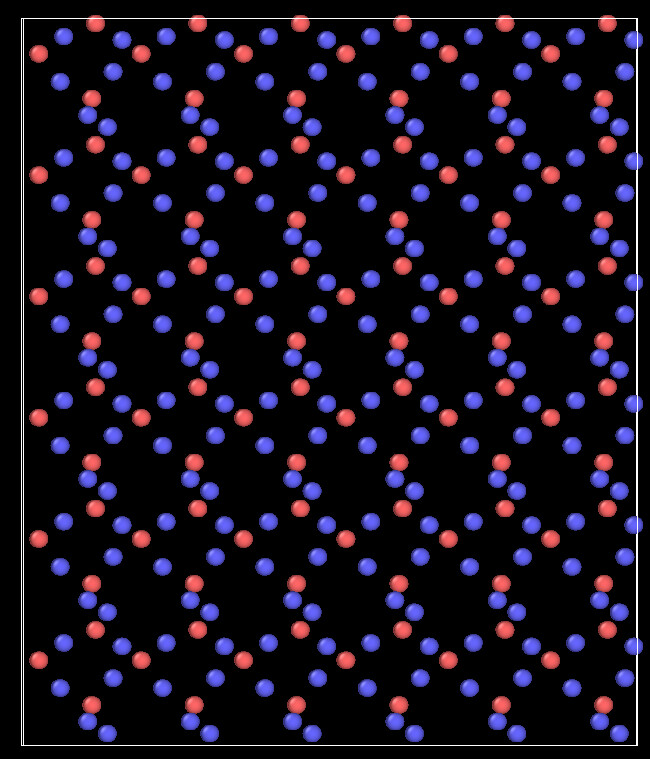

The code of second simulation:
variable num_dump_frames equal 100
variable num_particles equal 1944
variable temperature equal 300 # Initial state quantum temperature (K in metal units)
variable equilibration_steps equal 100000
variable production_steps equal 1000
variable pressure equal 0 # The pressure maintained by the NVT thermostat
variable cut_off equal 10 # Cut-off distance for the Buckingham term (Angstrom in metal units)
units metal
atom_style charge
timestep 1e-3
# variable lhalf equal ((${num_particles}/0.2)^(1/3))/2 # Aim for number density 0.1 at the begining.
variable lhalf equal ((33.26*${num_particles}/1.5)^(1/3))/2
region orthobox block -${lhalf} ${lhalf} -${lhalf} ${lhalf} -${lhalf} ${lhalf} units box
variable num_silicon equal ${num_particles}/3
variable num_oxygen equal ${num_particles}/3*2
create_box 2 orthobox
create_atoms 1 random ${num_silicon} 1234 orthobox
create_atoms 2 random ${num_oxygen} 4321 orthobox
mass 1 28.085500
mass 2 15.999400
set type 1 charge +2.4
set type 2 charge -1.2
pair_style hybrid/overlay buck/coul/long ${cut_off} table linear 39901
pair_coeff 1 1 buck/coul/long 0.0 1.0 0.0 #No interactions between Si atoms
pair_coeff 1 2 buck/coul/long 18003.757200 0.205205 133.538100
pair_coeff 2 2 buck/coul/long 1388.773000 0.362319 175.000000 #BKS interaction in PRL 64 1955 (1990)
pair_modify shift yes
pair_coeff 1 2 table potential_SiO2.TPF Si-O ${cut_off}
pair_coeff 2 2 table potential_SiO2.TPF O-O ${cut_off} #See the potential file for more information
kspace_style pppm 1.0e-4
##### Neighbor style
neighbor 3.0 bin
neigh_modify check yes every 1 delay 0 page 100000 one 10000
group Si type 1
group O type 2
# velocity all create ${temperature} 123
# fix f1 all npt temp ${temperature} ${temperature} 1 iso ${pressure} ${pressure} 1
variable varstep equal step
variable vartemp equal temp
variable varpress equal press
variable varetotal equal etotal
variable varlx equal lx
thermo 1000
thermo_style custom step temp press pe vol fnorm density
thermo_modify lost warn
minimize 0 1.0e-8 100000 10000000
fix f0 all box/relax aniso 1.0 vmax 0.001
minimize 0 1.0e-8 100000 10000000
unfix f0
minimize 0 1.0e-8 100000 10000000
reset_timestep 0
velocity all create ${temperature} 123
dump 1 all custom 1000 everything.dump id type x y z
fix f1 all npt temp ${temperature} ${temperature} 0.1 iso ${pressure} ${pressure} 1
restart 100000 TTM_pre_restart_*.mpiio
run 50000
unfix f1
fix f2 all npt temp ${temperature} 3000 0.1 iso ${pressure} ${pressure} 1
restart 100000 TTM_pre_restart_*.mpiio
run ${equilibration_steps}
unfix f2
fix f3 all npt temp 3000 3000 1 iso ${pressure} ${pressure} 1
restart 100000 TTM_pre_restart_*.mpiio
run 50000
unfix f3
fix f4 all npt temp 3000 ${temperature} 1 iso ${pressure} ${pressure} 1
restart 100000 TTM_pre_restart_*.mpiio
run ${equilibration_steps}
unfix f4
fix f5 all npt temp ${temperature} ${temperature} 1 iso ${pressure} ${pressure} 1
restart 50000 TTM_pre_restart_*.mpiio
run ${equilibration_steps}
unfix f5
Here the output:
LAMMPS (3 Mar 2020)
Created orthogonal box = (-17.5312 -17.5312 -17.5312) to (17.5312 17.5312 17.5312)
4 by 6 by 6 MPI processor grid
Created 648 atoms
create_atoms CPU = 0.00185729 secs
Created 1296 atoms
create_atoms CPU = 0.000839393 secs
Setting atom values ...
648 settings made for charge
Setting atom values ...
1296 settings made for charge
WARNING: 1 of 39901 force values in table are inconsistent with -dE/dr.
Should only be flagged at inflection points (../pair_table.cpp:483)
648 atoms in group Si
1296 atoms in group O
PPPM initialization ...
using 12-bit tables for long-range coulomb (../kspace.cpp:332)
G vector (1/distance) = 0.295882
grid = 27 27 27
stencil order = 5
estimated absolute RMS force accuracy = 0.00124108
estimated relative force accuracy = 8.61882e-05
using double precision KISS FFT
3d grid and FFT values/proc = 2016 175
Neighbor list info ...
update every 1 steps, delay 0 steps, check yes
max neighbors/atom: 10000, page size: 100000
master list distance cutoff = 13
ghost atom cutoff = 13
binsize = 6.5, bins = 6 6 6
2 neighbor lists, perpetual/occasional/extra = 2 0 0
Setting up cg style minimization ...
Unit style : metal
Current step : 0
Per MPI rank memory allocation (min/avg/max) = 5.38 | 5.387 | 5.389 Mbytes
Step Temp Press PotEng Volume Fnorm Density
0 0 3.2026541e+09 25819772 43104.96 3.9596915e+09 1.4998845
1000 0 -35715.755 -36722.935 43104.96 1.4719804 1.4998845
1418 0 -36527.461 -36736.989 43104.96 0.13020846 1.4998845
Loop time of 7.64799 on 144 procs for 1418 steps with 1944 atoms
94.5% CPU use with 144 MPI tasks x no OpenMP threads
Minimization stats:
Stopping criterion = linesearch alpha is zero
Energy initial, next-to-last, final =
25819772.0421 -36736.9894136 -36736.9894136
Force two-norm initial, final = 3.95969e+09 0.130208
Force max component initial, final = 2.78472e+09 0.0165201
Final line search alpha, max atom move = 4.76837e-07 7.87742e-09
Iterations, force evaluations = 1418 2404
Setting up cg style minimization ...
Unit style : metal
Current step : 1418
WARNING: Energy due to 3 extra global DOFs will be included in minimizer energies
Per MPI rank memory allocation (min/avg/max) = 5.379 | 5.387 | 5.389 Mbytes
Step Temp Press PotEng Volume Fnorm Density
1418 0 -36527.461 -36736.989 43104.96 0.13020846 1.4998845
1713 0 -146.94654 -36792.083 39084.805 3.6492124 1.6541585
Loop time of 11.5688 on 144 procs for 295 steps with 1944 atoms
98.7% CPU use with 144 MPI tasks x no OpenMP threads
Minimization stats:
Stopping criterion = linesearch alpha is zero
Energy initial, next-to-last, final =
-36736.9894136 -36792.0855389 -36792.0855389
Force two-norm initial, final = 1702.37 11.2702
Force max component initial, final = 999.002 10.5491
Final line search alpha, max atom move = 6.37916e-11 6.72941e-10
Iterations, force evaluations = 295 504
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 0.0065885 | 0.19125 | 0.35951 | 16.7 | 1.65
Kspace | 5.3458 | 5.7069 | 6.0823 | 4.1 | 49.33
Neigh | 6.4848e-05 | 0.0001456 | 0.00025173 | 0.0 | 0.00
Comm | 0.84002 | 1.2852 | 1.7779 | 10.3 | 11.11
Output | 0 | 0 | 0 | 0.0 | 0.00
Modify | 0 | 0 | 0 | 0.0 | 0.00
Other | | 4.385 | | | 37.91
Nlocal: 13.5 ave 25 max 0 min
Histogram: 2 6 12 10 32 13 38 11 15 5
Nghost: 1668.75 ave 1721 max 1627 min
Histogram: 5 12 22 27 22 21 16 13 5 1
Neighs: 3010.62 ave 6000 max 0 min
Histogram: 4 7 16 15 30 26 22 15 7 2
Total # of neighbors = 433530
Ave neighs/atom = 223.009
Neighbor list builds = 1
Dangerous builds = 0
PPPM initialization ...
using 12-bit tables for long-range coulomb (../kspace.cpp:332)
G vector (1/distance) = 0.29251
grid = 25 25 25
stencil order = 5
estimated absolute RMS force accuracy = 0.00159111
estimated relative force accuracy = 0.000110497
using double precision KISS FFT
3d grid and FFT values/proc = 2016 175
Neighbor list info ...
update every 1 steps, delay 0 steps, check yes
max neighbors/atom: 10000, page size: 100000
master list distance cutoff = 13
ghost atom cutoff = 13
binsize = 6.5, bins = 6 6 6
2 neighbor lists, perpetual/occasional/extra = 2 0 0
(1) pair buck/coul/long, perpetual
Setting up cg style minimization ...
Unit style : metal
Current step : 1713
Per MPI rank memory allocation (min/avg/max) = 5.354 | 5.368 | 5.382 Mbytes
Step Temp Press PotEng Volume Fnorm Density
1713 0 -146.81631 -36792.073 39084.805 3.6494501 1.6541585
2000 0 -26258.089 -36855.155 39084.805 1.5663468 1.6541585
2260 0 -28705.889 -36867.22 39084.805 0.1917403 1.6541585
Loop time of 1.95123 on 144 procs for 547 steps with 1944 atoms
93.2% CPU use with 144 MPI tasks x no OpenMP threads
Minimization stats:
Stopping criterion = linesearch alpha is zero
Energy initial, next-to-last, final =
-36792.0726676 -36867.2203252 -36867.2203252
Force two-norm initial, final = 3.64945 0.19174
Force max component initial, final = 0.275649 0.0279626
Final line search alpha, max atom move = 2.38419e-07 6.6668e-09
Iterations, force evaluations = 547 1094
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 0.0018582 | 0.40884 | 0.75569 | 22.7 | 20.95
Kspace | 0.7253 | 1.0696 | 1.4855 | 14.1 | 54.82
Neigh | 0.00023502 | 0.00078533 | 0.0012997 | 0.0 | 0.04
Comm | 0.34849 | 0.36513 | 0.37995 | 1.0 | 18.71
Output | 0.00012401 | 0.00012532 | 0.00014994 | 0.0 | 0.01
Modify | 0 | 0 | 0 | 0.0 | 0.00
Other | | 0.1067 | | | 5.47
Nlocal: 13.5 ave 26 max 0 min
Histogram: 3 7 8 24 15 33 29 13 10 2
Nghost: 1708.39 ave 1760 max 1672 min
Histogram: 5 14 25 36 17 19 17 4 6 1
Neighs: 3084.81 ave 6095 max 0 min
Histogram: 4 6 8 27 20 34 17 22 4 2
Total # of neighbors = 444212
Ave neighs/atom = 228.504
Neighbor list builds = 6
Dangerous builds = 0
PPPM initialization ...
using 12-bit tables for long-range coulomb (../kspace.cpp:332)
G vector (1/distance) = 0.29251
grid = 25 25 25
stencil order = 5
estimated absolute RMS force accuracy = 0.00159111
estimated relative force accuracy = 0.000110497
using double precision KISS FFT
3d grid and FFT values/proc = 2016 175
Neighbor list info ...
update every 1 steps, delay 0 steps, check yes
max neighbors/atom: 10000, page size: 100000
master list distance cutoff = 13
ghost atom cutoff = 13
binsize = 6.5, bins = 6 6 6
2 neighbor lists, perpetual/occasional/extra = 2 0 0
Setting up Verlet run ...
Unit style : metal
Current step : 0
Time step : 0.001
Per MPI rank memory allocation (min/avg/max) = 4.489 | 5.487 | 5.508 Mbytes
Step Temp Press PotEng Volume Fnorm Density
0 300 -26646.825 -36867.22 39084.805 0.1917403 1.6541585
1000 343.85313 -564.15007 -36972.811 28932.27 59.436285 2.2346142
2000 304.95356 -1025.1182 -37071.665 27916.943 55.230618 2.3158862
3000 279.44108 332.33376 -37092.667 27559.821 59.9403 2.3458956
...
48000 302.30967 -756.96403 -37172.927 26766.068 74.543831 2.4154636
49000 295.92801 1102.7683 -37173.712 26786.5 74.015064 2.4136212
50000 313.50318 -492.23162 -37170.384 26764.039 75.176159 2.4156467
Loop time of 96.9161 on 144 procs for 50000 steps with 1944 atoms
Performance: 44.575 ns/day, 0.538 hours/ns, 515.910 timesteps/s
90.7% CPU use with 144 MPI tasks x no OpenMP threads
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 6.2803 | 20.427 | 33.992 | 114.3 | 21.08
Kspace | 33.92 | 47.289 | 60.747 | 75.6 | 48.79
Neigh | 0.0044616 | 0.0092983 | 0.01304 | 1.8 | 0.01
Comm | 20.789 | 21.826 | 22.696 | 8.2 | 22.52
Output | 0.046474 | 0.04653 | 0.048118 | 0.1 | 0.05
Modify | 4.2353 | 5.3038 | 6.0309 | 20.1 | 5.47
Other | | 2.015 | | | 2.08
Nlocal: 13.5 ave 23 max 3 min
Histogram: 2 3 4 16 22 40 32 19 4 2
Nghost: 2313.69 ave 2359 max 2264 min
Histogram: 6 11 7 13 21 29 27 13 14 3
Neighs: 4489.42 ave 8648 max 1073 min
Histogram: 3 5 13 34 33 33 16 4 2 1
Total # of neighbors = 646476
Ave neighs/atom = 332.549
Neighbor list builds = 54
Dangerous builds = 0
PPPM initialization ...
using 12-bit tables for long-range coulomb (../kspace.cpp:332)
G vector (1/distance) = 0.299001
grid = 24 24 24
stencil order = 5
estimated absolute RMS force accuracy = 0.00130884
estimated relative force accuracy = 9.08938e-05
using double precision KISS FFT
3d grid and FFT values/proc = 1573 96
Neighbor list info ...
update every 1 steps, delay 0 steps, check yes
max neighbors/atom: 10000, page size: 100000
master list distance cutoff = 13
ghost atom cutoff = 13
binsize = 6.5, bins = 5 5 5
2 neighbor lists, perpetual/occasional/extra = 2 0 0
(1) pair buck/coul/long, perpetual
attributes: half, newton on
pair build: half/bin/atomonly/newton
stencil: half/bin/3d/newton
bin: standard
(2) pair table, perpetual, skip from (1)
attributes: half, newton on
pair build: skip
stencil: none
bin: none
Setting up Verlet run ...
Unit style : metal
Current step : 50000
Time step : 0.001
Per MPI rank memory allocation (min/avg/max) = 4.487 | 5.481 | 5.507 Mbytes
Step Temp Press PotEng Volume Fnorm Density
50000 313.50318 -506.25913 -37170.414 26764.039 75.176918 2.4156467
51000 325.43598 -1003.6766 -37165.932 26925.141 79.000562 2.4011931
52000 348.78456 -1960.7966 -37161.2 26673.295 79.863413 2.4238649
...
149000 3033.3578 8452.9713 -36675.97 25940.459 226.05507 2.4923407
150000 3007.8054 -1526.9646 -36640.964 26157.934 230.87087 2.4716196
Loop time of 188.901 on 144 procs for 100000 steps with 1944 atoms
Performance: 45.738 ns/day, 0.525 hours/ns, 529.379 timesteps/s
89.6% CPU use with 144 MPI tasks x no OpenMP threads
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 17.589 | 42.889 | 63.904 | 133.5 | 22.70
Kspace | 58.447 | 80.233 | 105.96 | 99.6 | 42.47
Neigh | 0.098466 | 0.1645 | 0.22195 | 5.8 | 0.09
Comm | 44.089 | 46.468 | 48.406 | 13.1 | 24.60
Output | 0.65792 | 0.65817 | 0.68351 | 0.3 | 0.35
Modify | 13.264 | 14.255 | 14.883 | 7.1 | 7.55
Other | | 4.233 | | | 2.24
Nlocal: 13.5 ave 19 max 4 min
Histogram: 1 1 4 2 26 15 41 20 25 9
Nghost: 2383.5 ave 2439 max 2346 min
Histogram: 8 15 19 30 28 21 14 7 0 2
Neighs: 4655.58 ave 7533 max 1167 min
Histogram: 1 2 6 20 28 32 26 20 7 2
Total # of neighbors = 670404
Ave neighs/atom = 344.858
Neighbor list builds = 876
Dangerous builds = 0
PPPM initialization ...
using 12-bit tables for long-range coulomb (../kspace.cpp:332)
G vector (1/distance) = 0.29957
grid = 24 24 24
stencil order = 5
estimated absolute RMS force accuracy = 0.00127928
estimated relative force accuracy = 8.88412e-05
using double precision KISS FFT
3d grid and FFT values/proc = 1573 96
Neighbor list info ...
update every 1 steps, delay 0 steps, check yes
max neighbors/atom: 10000, page size: 100000
master list distance cutoff = 13
ghost atom cutoff = 13
binsize = 6.5, bins = 5 5 5
2 neighbor lists, perpetual/occasional/extra = 2 0 0
(1) pair buck/coul/long, perpetual
attributes: half, newton on
pair build: half/bin/atomonly/newton
stencil: half/bin/3d/newton
bin: standard
(2) pair table, perpetual, skip from (1)
attributes: half, newton on
pair build: skip
stencil: none
bin: none
Setting up Verlet run ...
Unit style : metal
Current step : 150000
Time step : 0.001
Per MPI rank memory allocation (min/avg/max) = 4.499 | 5.483 | 5.507 Mbytes
Step Temp Press PotEng Volume Fnorm Density
150000 3007.8054 -1527.8513 -36640.966 26157.934 230.87082 2.4716196
151000 3064.7407 -7484.2784 -36668.281 26128.494 222.68537 2.4744045
152000 2950.1102 -830.59195 -36668.803 26240.596 227.46568 2.4638336
...
199000 3082.9038 -874.28135 -36688.239 25980.894 228.43904 2.4884618
200000 3054.2367 -1401.4104 -36720.677 25924.945 219.5275 2.4938322
Loop time of 94.5546 on 144 procs for 50000 steps with 1944 atoms
Performance: 45.688 ns/day, 0.525 hours/ns, 528.795 timesteps/s
89.6% CPU use with 144 MPI tasks x no OpenMP threads
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 11.504 | 21.741 | 29.614 | 77.9 | 22.99
Kspace | 31.784 | 39.567 | 50.931 | 60.0 | 41.85
Neigh | 0.10733 | 0.17659 | 0.22819 | 4.8 | 0.19
Comm | 22.229 | 23.503 | 24.558 | 9.1 | 24.86
Output | 0.44179 | 0.44486 | 0.46982 | 0.6 | 0.47
Modify | 6.5146 | 6.9706 | 7.3849 | 5.8 | 7.37
Other | | 2.152 | | | 2.28
Nlocal: 13.5 ave 20 max 4 min
Histogram: 1 0 2 8 16 50 36 14 15 2
Nghost: 2392.11 ave 2440 max 2357 min
Histogram: 8 15 17 16 42 20 16 5 3 2
Neighs: 4653.05 ave 7572 max 1519 min
Histogram: 1 3 9 21 35 28 29 7 9 2
Total # of neighbors = 670039
Ave neighs/atom = 344.67
Neighbor list builds = 943
Dangerous builds = 0
PPPM initialization ...
using 12-bit tables for long-range coulomb (../kspace.cpp:332)
G vector (1/distance) = 0.299793
grid = 24 24 24
stencil order = 5
estimated absolute RMS force accuracy = 0.00126793
estimated relative force accuracy = 8.80526e-05
using double precision KISS FFT
3d grid and FFT values/proc = 1573 96
Neighbor list info ...
update every 1 steps, delay 0 steps, check yes
max neighbors/atom: 10000, page size: 100000
master list distance cutoff = 13
ghost atom cutoff = 13
binsize = 6.5, bins = 5 5 5
2 neighbor lists, perpetual/occasional/extra = 2 0 0
(1) pair buck/coul/long, perpetual
attributes: half, newton on
pair build: half/bin/atomonly/newton
stencil: half/bin/3d/newton
bin: standard
(2) pair table, perpetual, skip from (1)
attributes: half, newton on
pair build: skip
stencil: none
bin: none
Setting up Verlet run ...
Unit style : metal
Current step : 200000
Time step : 0.001
Per MPI rank memory allocation (min/avg/max) = 4.499 | 5.483 | 5.508 Mbytes
Step Temp Press PotEng Volume Fnorm Density
200000 3054.2367 -1401.6824 -36720.678 25924.945 219.52749 2.4938322
201000 2931.7962 -484.79796 -36693.664 25950.465 227.35416 2.4913797
202000 2872.5191 479.89988 -36707.233 25637.049 226.87716 2.5218371
...
298000 395.28186 2002.2487 -37431.669 25434.452 84.723473 2.5419248
299000 373.13827 -1515.5951 -37439.278 25438.702 79.380137 2.5415
300000 333.59681 -539.66166 -37442.519 25517.819 80.161317 2.5336203
Loop time of 189.391 on 144 procs for 100000 steps with 1944 atoms
Performance: 45.620 ns/day, 0.526 hours/ns, 528.008 timesteps/s
89.6% CPU use with 144 MPI tasks x no OpenMP threads
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 24.942 | 44.044 | 62.168 | 104.8 | 23.26
Kspace | 59.963 | 79.133 | 100.18 | 80.6 | 41.78
Neigh | 0.099859 | 0.15363 | 0.19723 | 4.3 | 0.08
Comm | 44.382 | 47.198 | 49.083 | 13.3 | 24.92
Output | 0.89189 | 0.89253 | 0.93998 | 0.5 | 0.47
Modify | 12.878 | 13.854 | 14.631 | 8.3 | 7.32
Other | | 4.115 | | | 2.17
Nlocal: 13.5 ave 20 max 7 min
Histogram: 4 0 7 26 40 25 33 5 2 2
Nghost: 2420.05 ave 2463 max 2382 min
Histogram: 4 8 19 20 36 21 16 11 4 5
Neighs: 4723.28 ave 7485 max 2508 min
Histogram: 5 8 16 32 33 20 18 8 1 3
Total # of neighbors = 680153
Ave neighs/atom = 349.873
Neighbor list builds = 809
Dangerous builds = 0
PPPM initialization ...
using 12-bit tables for long-range coulomb (../kspace.cpp:332)
G vector (1/distance) = 0.300185
grid = 24 24 24
stencil order = 5
estimated absolute RMS force accuracy = 0.00124809
estimated relative force accuracy = 8.66753e-05
using double precision KISS FFT
3d grid and FFT values/proc = 1573 96
Neighbor list info ...
update every 1 steps, delay 0 steps, check yes
max neighbors/atom: 10000, page size: 100000
master list distance cutoff = 13
ghost atom cutoff = 13
binsize = 6.5, bins = 5 5 5
2 neighbor lists, perpetual/occasional/extra = 2 0 0
(1) pair buck/coul/long, perpetual
attributes: half, newton on
pair build: half/bin/atomonly/newton
stencil: half/bin/3d/newton
bin: standard
(2) pair table, perpetual, skip from (1)
attributes: half, newton on
pair build: skip
stencil: none
bin: none
Setting up Verlet run ...
Unit style : metal
Current step : 300000
Time step : 0.001
Per MPI rank memory allocation (min/avg/max) = 4.499 | 5.484 | 5.508 Mbytes
Step Temp Press PotEng Volume Fnorm Density
300000 333.59681 -540.60146 -37442.521 25517.819 80.161311 2.5336203
301000 329.53265 2093.002 -37447.358 25461.899 78.231673 2.5391846
302000 320.67692 -1505.3053 -37452.013 25454.302 75.217439 2.5399425
...
398000 302.96029 12.910631 -37452.992 25495.397 75.360814 2.5358485
399000 309.50624 2286.0348 -37453.572 25442.566 75.253949 2.541114
400000 302.62384 -2668.8727 -37454.963 25502.651 75.12892 2.5351271
Loop time of 186.067 on 144 procs for 100000 steps with 1944 atoms
Performance: 46.435 ns/day, 0.517 hours/ns, 537.442 timesteps/s
89.4% CPU use with 144 MPI tasks x no OpenMP threads
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total
---------------------------------------------------------------
Pair | 24.963 | 43.917 | 66.592 | 118.1 | 23.60
Kspace | 53.52 | 76.914 | 98.403 | 90.9 | 41.34
Neigh | 0.0002706 | 0.0004136 | 0.00058636 | 0.0 | 0.00
Comm | 43.646 | 46.248 | 48.319 | 14.4 | 24.86
Output | 1.9064 | 1.9068 | 1.9264 | 0.2 | 1.02
Modify | 12.007 | 13.039 | 13.663 | 8.3 | 7.01
Other | | 4.042 | | | 2.17
Nlocal: 13.5 ave 19 max 7 min
Histogram: 2 1 7 10 17 72 14 10 6 5
Nghost: 2419.74 ave 2454 max 2397 min
Histogram: 9 12 32 27 19 24 10 5 3 3
Neighs: 4728.12 ave 7434 max 2645 min
Histogram: 6 5 19 40 27 23 12 6 3 3
Total # of neighbors = 680849
Ave neighs/atom = 350.231
Neighbor list builds = 2
Dangerous builds = 0
System init for write_data ...
PPPM initialization ...
using 12-bit tables for long-range coulomb (../kspace.cpp:332)
G vector (1/distance) = 0.3002
grid = 24 24 24
stencil order = 5
estimated absolute RMS force accuracy = 0.00124735
estimated relative force accuracy = 8.6624e-05
using double precision KISS FFT
3d grid and FFT values/proc = 1573 96
Neighbor list info ...
update every 1 steps, delay 0 steps, check yes
max neighbors/atom: 10000, page size: 100000
master list distance cutoff = 13
ghost atom cutoff = 13
binsize = 6.5, bins = 5 5 5
2 neighbor lists, perpetual/occasional/extra = 2 0 0
(1) pair buck/coul/long, perpetual
attributes: half, newton on
pair build: half/bin/atomonly/newton
stencil: half/bin/3d/newton
bin: standard
(2) pair table, perpetual, skip from (1)
attributes: half, newton on
pair build: skip
stencil: none
bin: none
Total wall time: 0:13:00