For the basic.cmake, I just modified the following line:
set(ALL_PACKAGES GPU KOKKOS KSPACE MANYBODY MOLECULE REAXFF)
The following command was used to run the simulation:
mpirun -np 2 …/…/lmp_kokkos -k on g 2 -sf kk -pk kokkos newton on neigh half binsize 6.0 -in in.test
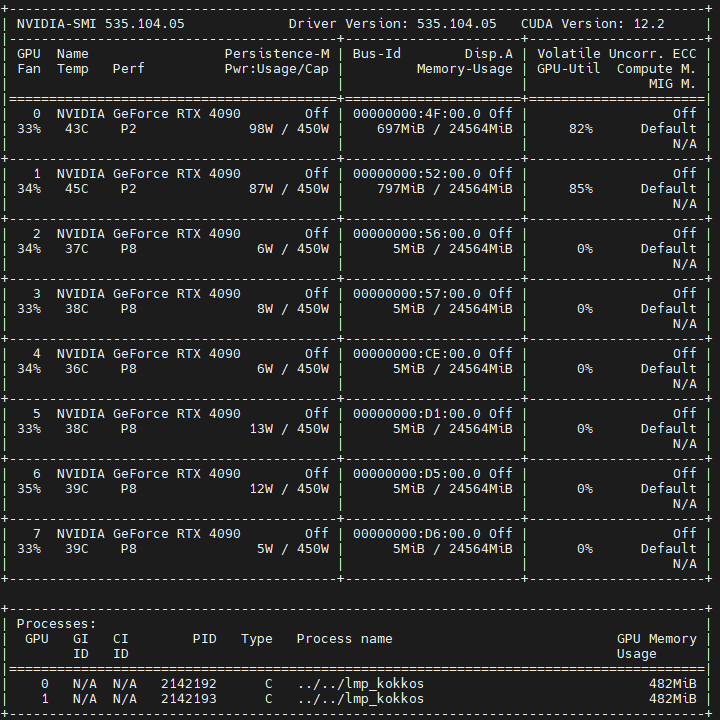
For 2 GPUs, the GPU Util is about 80%. I think it’s reasonable.
When I use 4 or 8 GPUs, the Util is 0%-1%, which is so strange and slow.
The input file and log.lammps for 2 GPUs case can be seen attached.
Is there someone knows the reason? Thanks so much.
6100 atoms is nowhere near enough to saturate a GPU. im surprised you’re even getting two GPUs to 80%.
from previous discussion with @stamoor, REAXFF can do 100k - 1M atoms per V100 for optimal performance.
ADA cards have 2816-18176 cores per GPU [NVIDIA Ada Lovelace GPUs | PNY Pro | pny.com], so when spreading 6100 atoms across 8 GPUs (762 atoms per GPU) there’s almost no floating point computations happening. All LAMMPS/KOKKOS is doing is GPU-CPU memory bandwidth, and network communication between MPI ranks.
you need a lot more reaxff atoms to see the scaling you’re expecting.
you have two more problems:
(1) you need to compile a CUDA-aware openmpi:
WARNING: Turning off GPU-aware MPI since it is not detected, use ‘-pk kokkos gpu/aware on’ to override (src/KOKKOS/kokkos.cpp:291)
using 1 OpenMP thread(s) per MPI task
(2) this one im not familar with but i would fix that also
WARNING: Van der Waals parameters for element LI indicate inner wall+shielding, but earlier atoms indicate a different van der Waals method. This may cause division-by-zero errors. Keeping van der Waals setting for earlier atoms. (src/REAXFF/reaxff_ffield.cpp:249)
WARNING: Van der Waals parameters for element AL indicate inner wall+shielding, but earlier atoms indicate a different van der Waals method. This may cause division-by-zero errors. Keeping van der Waals setting for earlier atoms. (src/REAXFF/reaxff_ffield.cpp:249)
ps: you dont need -D Kokkos_ENABLE_OPENMP=yes either if you’re using GPUs
According to the manual, I added the following lines to the environment for binding GPUs and MPI.
export OMP_PROC_BIND=spread
export OMP_PLACES=threads
the log file for 4 and 8 gpus is the same as the log of 2 gpus, as can be seen attachments. log.lammps-4gpus (4.0 KB) log.lammps-8gpus (4.0 KB)
BTW, I also tesed the system with 10,890 atoms. The issue is the same.
100000 atoms across 8 GPUs is still almost nothing. Try 1M, 10M, …
also you have both NVT and NPT fixes running at the same time. i have no idea what kind of shenanigans that might cause.
fix 1 all nvt/kk temp 300 1000 $(100*dt) drag 1
fix 2 all npt/kk temp 1000.0 1000.0 $(100*dt) iso 0 0 $(1000*dt)
WARNING: One or more atoms are time integrated more than once (src/modify.cpp:296)
and compute peratom all pe/atom which doesnt have a kokkos version so that’s a lot of gpu-cpu memory bandwidth at every timestep
Thanks for your suggestions.
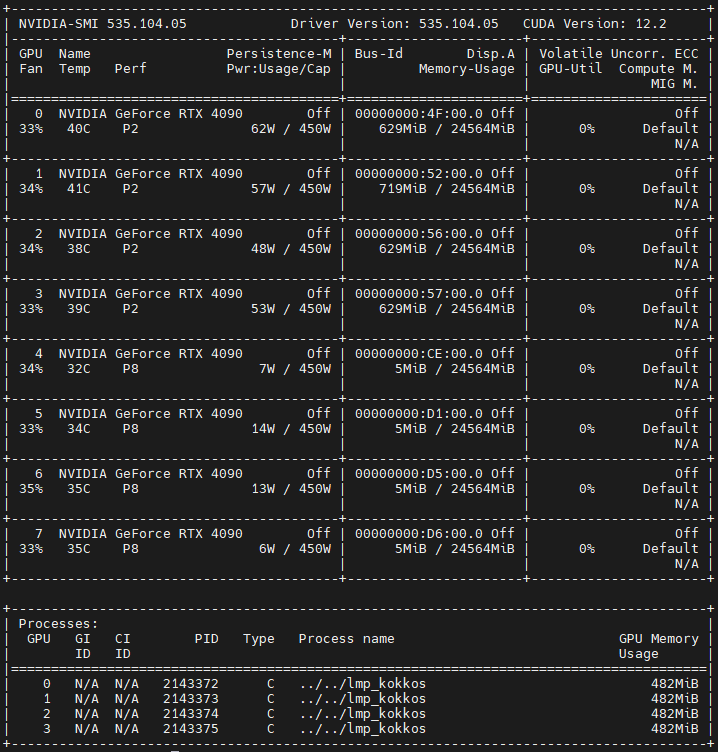
After removed the “compute peeratom all pe/atom” and modifying the two fix, I have tested the system with 1M and 2M by using 3, 4, 8 GPUs.
2M system:
As you said, the GPU Utils increase a little bit for large system, about 3 GPUs (mostly: 50%) is larger than that of 4 GPUs (mostly: 10%) and 8 GPUs (mostly: 5%).
BTW: If I use 2 GPUs for 1M system, the GPU Utils is larger than 80%.
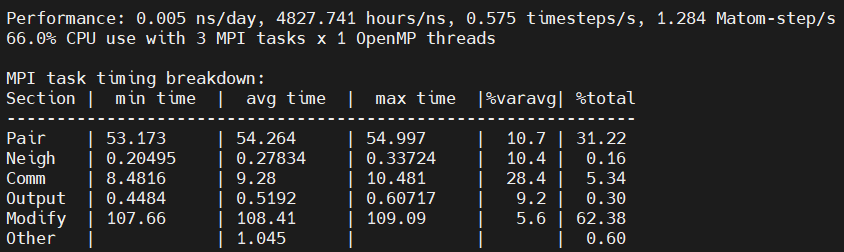
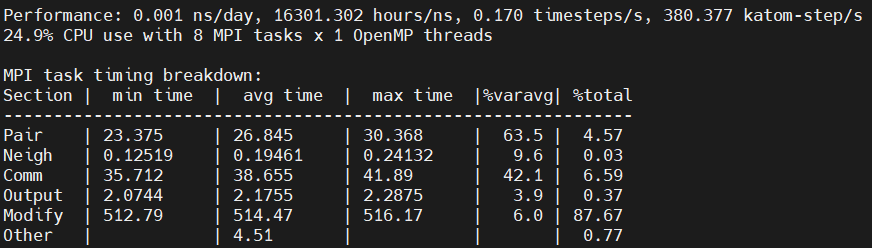
Another question: Why the “Modify” part take a lot of time? This part will increase with increasing the number of GPU part, as can be seen below:
modify are the fixes in LAMMPS (npt and qeq in your case). @stamoor is the expert on reaxff and he would know a lot more than me about why the fixes are computationally blowing up in a reaxff context. im guessing something to do with charge equilibration.
@Withyou can you please use export CUDA_LAUNCH_BLOCKING=1 to get an accurate timing breakdown at the end of the log file for the KOKKOS package, so we know where the time is increasing?
I would focus more on LAMMPS performance than on GPU utilization since that is what you really care about. Looking at your logfile output, the “Performance” in katom-steps/s drops when you add more GPUs, which means that either you don’t have enough work to keep all the GPUs busy, or you have some other problem.
As Mitch said, you really need to use CUDA-aware MPI to get good performance. It is being turned off by LAMMPS since it isn’t supported by your MPI library:
WARNING: Turning off GPU-aware MPI since it is not detected, use ‘-pk kokkos gpu/aware on’ to override (src/KOKKOS/kokkos.cpp:291)
You need to fix this warning. Turning it off manually using “gpu/aware off” gives the same (bad) performance since your MPI doesn’t support it and LAMMPS already turned it off by default. So it is off in all cases in your tests.
Thanks very very much for your and Mitch’s @alphataubio suggestions.
After fixing the warning of “gpu/aware off”, I think the performance for 8-GPUs is reasonable, as can be seen below and the attachments. Now, the GPUs Utils is mostly about 70-80%.
You can ignore that warning. It means some code (for minimize only) isn’t running on the GPU but minimize typically isn’t the bottleneck in a simulation. The only way to fix the warning is for a developer to convert more of the minimize code to use Kokkos.
it’s because minimize/kk is missing some device code to pack/unpack/sort data on the device when reneighboring occurs. however this only happens every ~100 steps and typically the minimize phase of a simulation is 1-5% of overall running time. i looked at it but the minimize packing/sorting code is not trivial to port to KOKKOS like almost everything else in LAMMPS. even if you manage to make it 10X faster, you’d get less than 1% improvement in overall simulation running time.
However one thing you can do to squeeze more performance out of KOKKOS now that you have a cuda-aware openmpi is to use 2:1 mpi ranks to gpu devices, ie. with 8 gpus you need mpirun -np 16 ...
here’s the logic: gpus are extremely fast to compute, so fast that one cpu is not feeding data fast enough to the gpu to keep it 100% busy. because of the asynchronous cpu-gpu memory transfers vs computation on the gpu, one cpu is feeding data to the gpu while the gpu is computing what it got from the other cpu. however going to 4:1 ratio doesnt help much, and going 8:1 ratio actually goes backwards because now there’s too much network overhead from the domain decomposition across too many mpi ranks.
Thanks so much for your explaniation.
Yeah, I agree that the total performance increase a little bit after fixing minimize/kk.
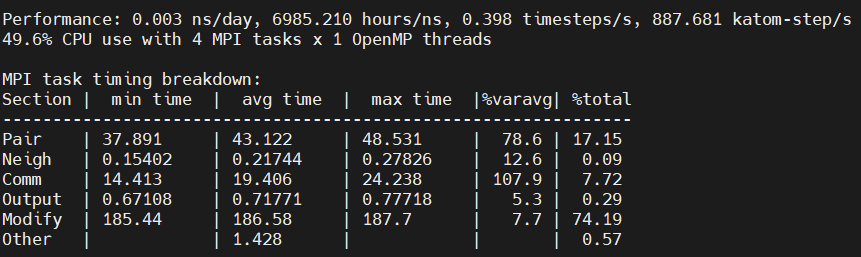
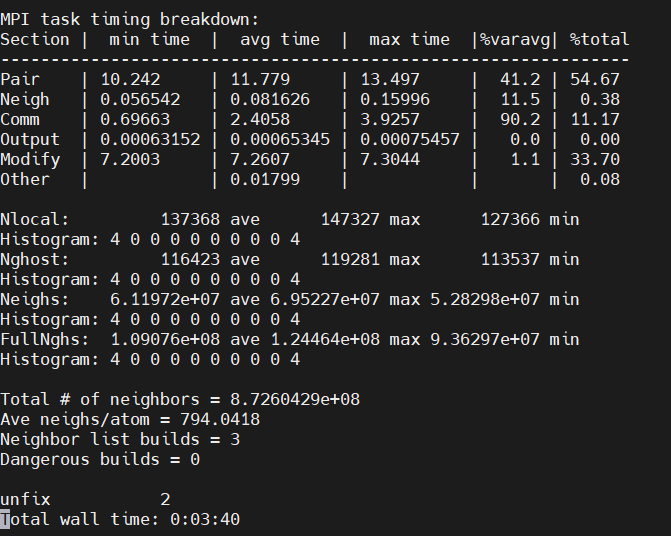
For your second suggestion, I understand that. Howerver, the simulation for 2M system is not good by using 16 mpi tasks , as can be seen the logs. The modify time increase a lot from 8 mpi (31.93) to 16 mpi (56.81), instead of Comm time.
I am wondering that the manual always suggests using the same number of mpi tasks and gpus: " Typically the -np setting of the mpirun command should set the number of MPI tasks/node to be equal to the number of physical GPUs on the node."