Hello,
I was trying to simulate grain boundaries in a system containing 5 metals with defined number of moles. But the pressure of my system become ‘nan’ in the second minimization step.
The code is given below.
Hello,
I was trying to simulate grain boundaries in a system containing 5 metals with defined number of moles. But the pressure of my system become ‘nan’ in the second minimization step.
The code is given below.
the first thing to check when getting NaN for energies, forces, or pressure is for overlapping atoms. You are creating atoms in adjacent regions with different lattices.
you seem to be having 6 atoms closer than 0.01 \AA and 6 more atoms within less than 0.2 \AA of each other.
axel.
Thank you for the constant and timely support Dr. Kohlmeyer. By taking your suggestion I deleted atoms and the pressure is not ‘nan’ anymore.
I am facing another problem in the same code. I am creating atoms twice in two different regions- 64000 at one time then 64760 again.
According to my system I would like to allocate the following number of atoms -
Mo - 103974 atoms (0.8075 mole)
W- 5472 atoms (0.0425 mole)
Ta- 12876 atoms (0.1 mole)
Ti - 3219 atoms (0.025 mole)
Zr-3219 atoms (0.025 mole)
I tried the following code -
clear
units metal
dimension 3
boundary p p p
atom_style atomic
lattice bcc 3.21
region whole block 0 128 0 128 0 128 units box
create_box 5 whole
region upper block INF INF 64 128 INF INF units box
lattice bcc 3.21 orient x 0 3 1 orient y 0 -1 3 orient z 1 0 0
create_atoms 1 region upper
region lower block INF INF 0 64 INF INF units box
lattice bcc 3.21 orient x 0 3 -1 orient y 0 1 3 orient z 1 0 0
create_atoms 2 region lower
variable fa equal 24786
variable fb equal 19314
variable fc equal 6438
variable fd equal 3219
variable ft equal 128760
set type 1 type/fraction 2 (v_fa/v_ft) 1734536
set type 2 type/fraction 3 (v_fb/(24786)) 1734535
set type 3 type/fraction 4 (v_fc/(19314)) 1734534
set type 4 type/fraction 5 (v_fd/(6438)) 1734533
group Mo type 1
group W type 2
group Ta type 3
group Ti type 4
group Zr type 5
But I am getting the following output
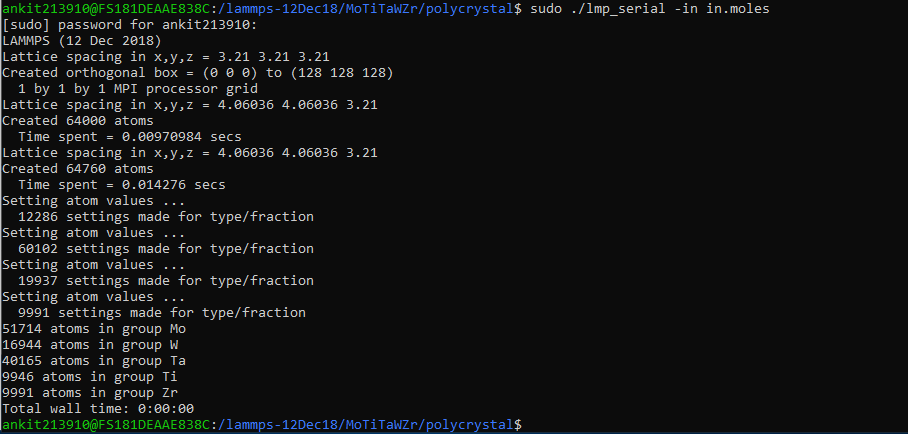
The number of atoms are not that which I am trying to get.
Could you kindly look into this and suggest me where I am going wrong?
[…]
The number of atoms are not that which I am trying to get.
Could you kindly look into this and suggest me where I am going wrong?
this is a simple exercise in calculus, reading the documentation, and careful observation
axel.
p.s.: i am appalled to see that you are using “sudo” to run simulations. using sudo this liberally is a very, very bad habit. LAMMPS is not designed to be run as superuser and can render your installation unusable, if used carelessly. for user-level applications like LAMMPS there should be no need to use superuser privilege to compile and use it.
Thanks Dr. Kohlmeyer. I was able to correct the simulation after reading the documentation.
I was having to use the sudo as lammps was not running and was denying permission to run. I will get it fixed Dr. Kohlmeyer. Thank you for correcting me on that.
Sincerely,
Ankit Roy
Thanks Dr. Kohlmeyer. I was able to correct the simulation after reading the documentation.
I was having to use the sudo as lammps was not running and was denying permission to run.
that usually happens when people get an error and then prefix a command with sudo in the hope to resolve this error. but this usually does not resolve the error. it can change permissions on generated files, though, and from then on you will get permission denied errors unless you always use sudo. this is one of the reasons why the liberal and careless use of sudo has been proliferating so much up to the point that people think that using sudo can fix all kinds of problems (which it can not).
axel.
Alright Dr. Kohlmeyer. I will look into this issue and get it resolved. Thank you for the constant guidance.