Dear lammps users,
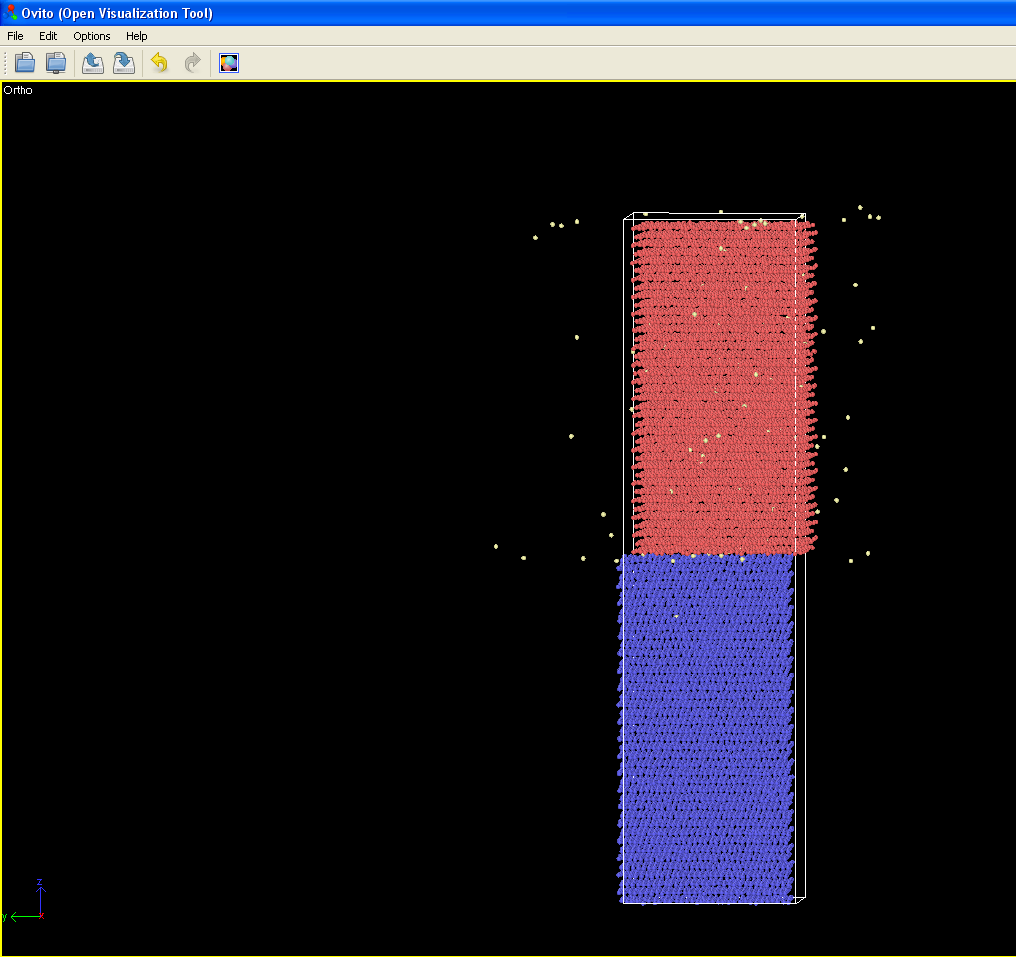
I am trying to simulate sigma 5(310) grain boundary of Ni with randomly distributed hydrogen atoms. I am trying to calculate diffusion coefficients at temperatures above 500K. I have minimized the structure using nve and box relaxc aniso. after equilibration, I am using nve to calculate diffusion coefficients. My question is when I am using nve I see the upper part of grain is sliding on lower as shown in attached picture and this effect is incresing with temperature. How can keep the structure stable without sliding?
Grainboundary.bmp (2.78 MB)
{kind=link}