To the LAMMPS community,
Using LAMMPS 64-bit 20160216 on a Windows 10 computer.
As part of my research project, I am running a simulation involving the
compression of a sheet of single layer graphene. As far as I know, I have
the needed elements included in my script to compress two sides of the
sheet to produce a bended configuration, like a sheet of paper, on the
center region of the sheet, but there is no movement of the atoms at all. I
have tried different boundary conditions, different means to move a group
of atoms, and different ‘fix’ conditions to satisfy thermodynamics with no
avail.
stephen,
i had a closer look at your input and there is quite a long list of issues
that need changing. it looks to me that you are very much in need of
consulting with somebody that had done such kind of studies before, because
there are a large number of obvious but also subtle things that need to be
considered and tested carefully, or else you'll be confronted with the
merciless GI-GO properties of MD simulations.
i am going over issues that i found when doing a quick glance and making
some changes to come up with a minimal template that can then be further
refined.
# 1: your input is lacking the final newline (you used notepad, right?).
LAMMPS may not read the last line because of that. better to use a proper
text editor or add a few empty lines at the end.
log log_in_test.txt
package omp 64
#2: 64 threads is ridiculous and overkill. multi-threading rarely scales
that far, and particularly not for the input below, as it contains "dump
image", which is not multi-threaded at all. with as frequent as you output
images, it will be hard to get any significant speedup from threads beyond,
say, 5x.
# ----- Simulation Setup -----
units metal
atom_style molecular
dimension 3
boundary p p p
#3: if you want deformation so that the sheet bends in Z, you should not
have periodic boundaries in Z. best in this case is to use "boundary p p m"
instead
newton on
# ----- Create Atoms -----
read_data data_graphene_5x5.txt
#4: i already mentioned that for AIREBO you must not define any bonded
interactions. "special_bonds lj/coul 1.0 1.0 1.0" serves as a workaround
here.
region 1 block &
INF 0.0 &
INF INF &
INF INF
group left region 1
region 2 block &
48.0 INF &
INF INF &
INF INF
group right region 2
group sheet subtract all left right
group contact union left sheet right
#5: groups/regions are not really needed for what you want to do. with PBC,
you have to deform the box, otherwise you compress and stretch at the same
time due to PBC.
# ----- Interatomic Potentials -----
pair_style airebo/omp 1.5
#6: why 1.5? the recommended default is 3.0 and before you have a working
simulation, you should not mess with this.
pair_coeff * * CH.airebo C
bond_style harmonic/omp
bond_coeff 1 25.0 1.421
angle_style harmonic/omp
angle_coeff 1 85.0 120.0
dihedral_style harmonic/omp
dihedral_coeff 1 1.6 1 2
#7: get rid of those styles and coefficients for bonded interactions. see
#4.
# ----- Processor Behavior -----
neighbor 2.0 bin
neigh_modify delay 10 &
every 10 &
check yes &
once yes &
cluster yes &
include all &
exclude none &
page 1000000 &
one 10000
#8: this is a mess too. you should just set the minimum amount of
flags. "neigh_modify
delay 0 every 1 check yes" should do for starters
# ----- Simulation Constraints -----
# fix 1 all nvt/omp &
# temp 4.0 4.0 10.0
#9: if you want to see displacement, you *have* to do time integration,
however nvt is not a good idea (what does the sheet exchange kinetic energy
with. it would be sufficient to set an initial amount of kinetic energy,
and then using "fix nve".
displace_atoms left move &
5.0 0.0 0.0
#10: displace_atoms is a one-time operation. that is not what you want.
"fix move" would be the way to do that continuously during a run, but - as
explained above - atom displacements are not a good idea with a periodic
system.
#11: more importantly, before doing any kind of simulation, you will have
to relax your system. visualization in VMD with periodic display enabled
(or just looking at the pressure of a run 0) shows that your cell
dimensions are not perfect, and also the optimal atom-atom distances vary
by potential. so you may have a very high potential energy structure, that
might just "explode" when doing time integration. instead doing a
minimization is necessary.
#12: since after the minimization with fixed volume, there is still
significant residual pressure, as second minimization with box adjustments
is required.
#13: at this point, you might want to start an MD, but before doing any
complex operation, you will need to assign some kinetic energy and do a bit
of an equilibration. if you don't, you atoms will have no velocities at all
and since there will be no forces in z direction due to all atoms being
perfectly in the same plane, it cannot bend.
# ----- Create Output Files -----
dump 1 all image 10 image_test_*.jpg &
type &
type &
size 1000 1000
#14: as mentioned before, dumping images every 10 steps is a bit excessive
and slowing down your run. also using dump movie is usually providing more
valuable information about dynamics.
# ----- Run the Simulation -----
timestep 1
#15: the time unit in metal units is a picosecond. 1ps is waaaay too much
for stable time integration. 1fs is more it (since you have no hydrogens,
otherwise 0.25 might be more adequate).
thermo 10
thermo_style custom &
step &
pe &
lx ly lz
run 100
#16: 100 steps is very short. remember that 1 step is a femtosecond and a
significant deformation over 100fs is a *massive* shock to the system. i've
changed this to 10000 in my modified input, which is still very fast, but
this is mainly to show the principle anyway.
#17: the way to impose the compression is to use fix deform. due to using
dump image/movie, i've compressed the box in y direction for better
visualization
#18. you probably want to continue the simulation with some additional
equilibration after the deformation is done. just unfix the deform fix.
I have included a copy of the graphene data file created by VMD 1.9.2 and
a copy of the script I have been testing.
I would appreciate any insight as to what I am not taking into account for
this simulation.
please note, that this is just a crude check of mostly technical issues.
the resulting input is not likely to be proper for any kind of real
production simulations that would provide meaningful data. it merely
demonstrates the principal workflow. many factors will have impact on the
result, particularly finite size effects.
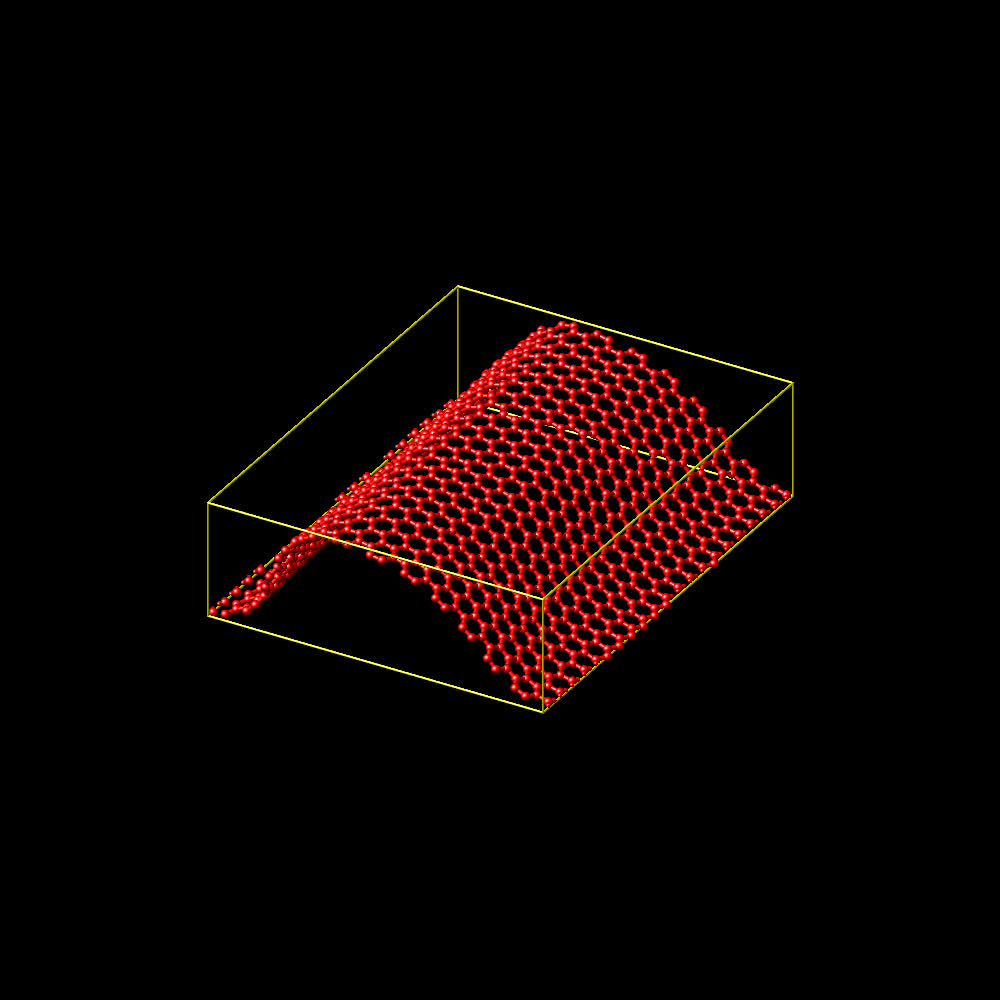
i am attaching a picture of the final configuration and the fully modified
input workflow demo.
axel.
in_test-ak.txt (1.3 KB)