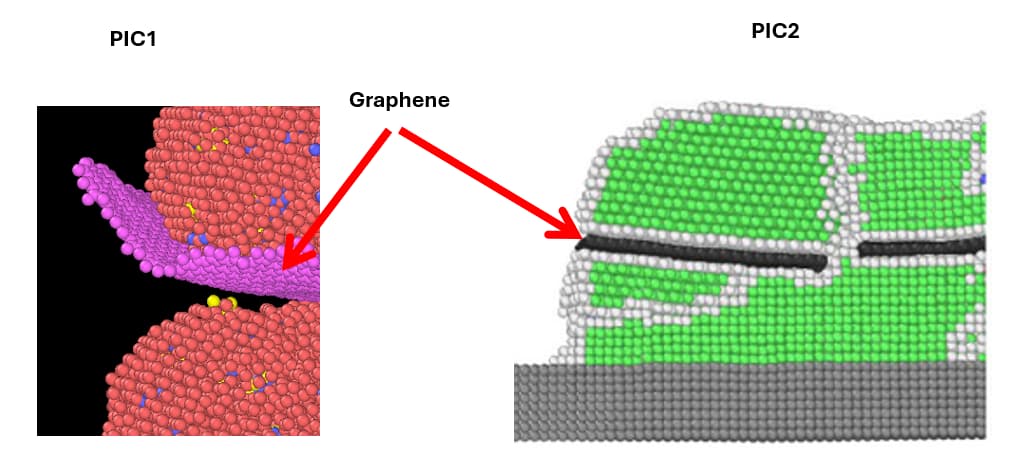
I am simulating a graphene sheet sandwiched between Al alloy particles using the following code.
dimension 3
units metal
boundary p p p
atom_style atomic
atom_modify map array
comm_modify cutoff 5.25
variable sixth equal “1/6”
variable twothirds equal “2/3”
variable alattice equal “2.4595”
variable a1lattice equal “1”
variable a2lattice equal “1.73203”
lattice custom {alattice} a1 {a1lattice} 0 0 a2 0 {a2lattice} 0 basis 0 0 0 basis 0.5 {sixth} 0 basis 0.5 0.5 0 basis 0 ${twothirds} 0
variable boxside equal 200
variable xside equal “round(v_boxside/v_a1lattice/v_alattice/2)” #round ensures that we get complete unit cells
variable yside equal “round(v_boxside/v_a2lattice/v_alattice/2)”
region box block -{xside} {xside} -{yside} {yside} -3 3 units lattice
region graphene block -{xside} {xside} -{yside} {yside} 0 0.1 units lattice
create_box 4 box
create_atoms 4 region graphene
mass * 12.011
The issue is that when I increase the temperature of the system, the graphene sheets curl up, as shown in pic1. I expected them to look like in pic2 after the alloy melts.
Any suggestion is highly appreciated.