Hello everyone,
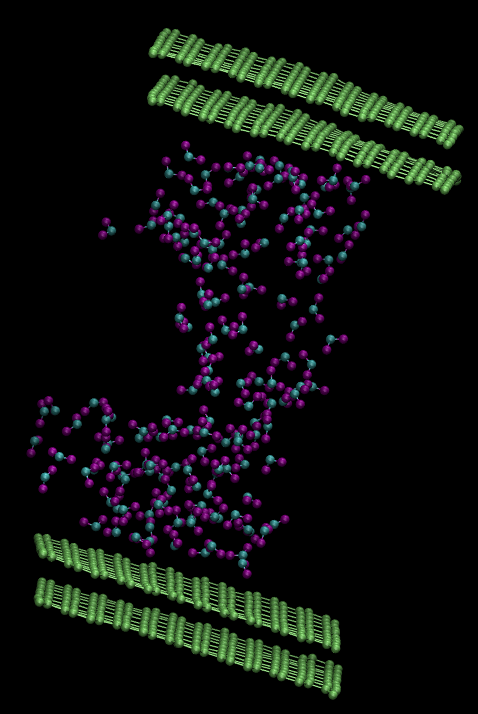
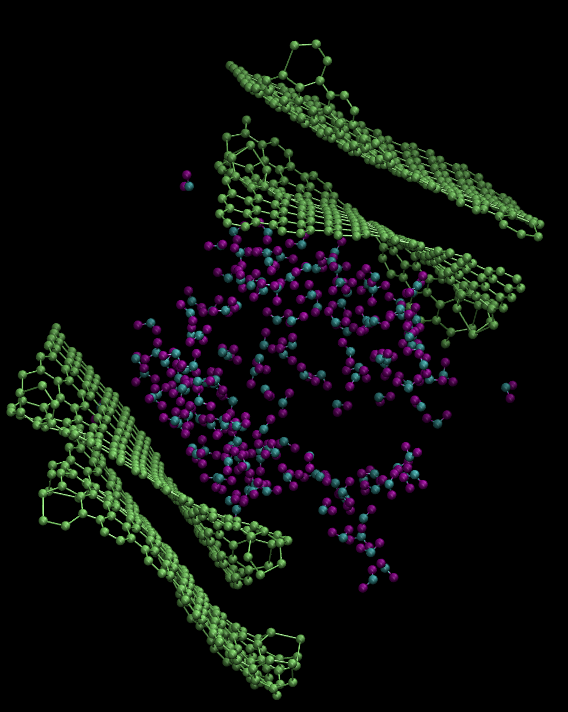
I have built a geometry of water (PACKMOL) and graphene (VMD Nanobuilder) together in a simulation box with LAMMPS and read the data with VMD’s Tk-console. I have two layers of graphene on top and bottom of the box with water sandwiched between them. I put 2 angstrom of vacuum between the water-graphene interfaces and 3 angstrom vacuum at the bottom and top of the box. I tested two cases. Firstly, I did not put any vacuum in the x and y directions. After equilibrating the system, the graphene sheets are found to be rolled across the y axis. Secondly, I put 3 angstrom vacuum in x and y directions also. This time, the graphene sheets seemed to be stable but water molecules are traveling into the vacuum in x and y directions. I checked the box dimensions and the highest and lowest coordinates of the atoms in the system. The coordinates of the atoms are contained in the simulation box. I am giving you two images of my system.. First image is my first test. Second image is my second test. I will greatly appreciate any suggestion or direction. Here is my input file:
water with graphene
echo screen
log debug.log
units real
atom_style full
bond_style harmonic
angle_style harmonic
dihedral_style harmonic
improper_style cvff
pair_style lj/cut/coul/long 10
kspace_style ewald 1.0e-6
neighbor 2.0 bin
#neigh_modify delay 0 every 1 one 10000 check yes
neigh_modify delay 0 every 1 check yes
boundary p p p
read_data data.waterlong
pair_modify mix arithmetic tail yes
group graphene type 3
group water type 1 2
group oxygen type 2
group hydrogen type 1
set group oxygen charge -0.8476
set group hydrogen charge 0.4238
#pair_coeff 3 3 0.4396 3.851
pair_coeff 3 3 0.12 3.2963
pair_coeff 2 2 0.15535 3.166
#pair_coeff 2 3 0.392 3.19
pair_coeff 2 3 0.095 3.19
pair_coeff 1 1 0.0 0.0
pair_coeff 1 2 0.0 0.0
#pair_coeff 1 3 0.129 2.801
pair_coeff 1 3 0.0 0.0
bond_coeff 2 469 1.4
bond_coeff 1 554.135 1.000
angle_coeff 2 85 120
angle_coeff 1 45.7696 109.47
dihedral_coeff 1 12 -1 2
improper_coeff 1 5.3 -1 2
#improper_coeff 1 3 1 2
compute tmpall all temp
compute water_temp water temp
compute graphene_temp graphene temp
compute water_comtemp water temp/com
minimize 1.0e-15 1.0e-15 1000 100000
velocity water create 298.0 12345 temp water_temp
velocity graphene create 298.0 12345 temp graphene_temp
#fix 1 water shake 1.0e-8 100 0 b 1 a 1
#fix 2 graphene setforce 0.0 0.0 0.0
#fix 3 all nvt temp 300.0 300.0 100.0
#fix 3 all nve
#fix 3 all npt temp 300.0 300.0 100.0 iso 1.01325 1.01325 100
fix 2 water nvt temp 298.0 298.0 5.0
fix_modify 2 temp water_comtemp
fix 3 graphene nvt temp 298.0 298.0 5.0
fix_modify 3 temp graphene_temp
fix 4 graphene momentum 1 linear 1 1 1
fix 5 water shake 0.001 20 1000000 b 1 a 1
#fix 6 all box/relax iso 1.01325
timestep 0.01
thermo 50
thermo_style one
thermo_modify lost error flush yes
dump 1 all xyz 1 graphene1.xyz
dump_modify 1 element C H O
run 100000
write_data data.waterlong_equil pair ij
Best Regards,
Baig Abdullah Al Muhit
PhD student/Graduate Teaching Assistant
Department of Civil and Environmental Engineering
School of Engineering
Vanderbilt University, Nashville, TN