While simulating grains whose diameter grows with time at each timestep, I did not manage to concomitantly adapt with time the cutoff value of their pair interaction.
The value of the cutoff set in the pair_coeff command is not re-evaluated at each time step, and it seems that the fix adapt command cannot be applied to pair_style granular.
A brutal solution could consist of setting a cutoff large enough to accommodate grain neighbor detection at their maximal size, but this would dramatically increase the computation time when many particles are growing together.
Is there a solution to continuously increase the neighbor detection cutoff at each timestep for granular simulations?
Thank you very much for your assistance,
Best regards,
Samuel
Below is an illustrating example code of two neighboring grains growing and pushing each other until the neighbor detection fails and they start overlapping.
# pair_grain_growth.lammps
# 1) Initialization
dimension 3
atom_style sphere 1
units si
boundary f f f
comm_modify vel yes
newton off
atom_modify map yes
# 2) Parameters
variable grain_diam equal 0.02 #[m]
variable Kn equal 50 #[N/m]
variable dissip equal 1 #[1/(m.s)]
variable muFrict equal 0.1
variable growth_ratio equal 10
# 3) Geometric parameters and regions
variable box_size equal v_growth_ratio*v_grain_diam*5
region simulation_box block -${box_size} ${box_size} -${box_size} ${box_size} -${box_size} ${box_size} units box
create_box 1 simulation_box
# 4) Create grains
variable init_sep equal v_grain_diam/2
create_atoms 1 single 0 -${init_sep} 0
create_atoms 1 single 0 ${init_sep} 0
set type 1 diameter ${grain_diam}
set type 1 density 1
compute compdiam all property/atom diameter radius
compute grainforces all property/atom fx fy fz
# 6) fix forces and dynamics
variable thermo_num equal 1000
thermo ${thermo_num}
thermo_style custom step etotal
fix damp all viscous 0.01
fix mynve all nve/sphere
fix nvelim all nve/limit ${grain_diam}
variable set_timestep equal 0.00001
timestep ${set_timestep}
# 7) Growth
variable radgrowth equal 1 # growth rate in diameter unit per s
variable diamtime equal v_grain_diam*(1+v_radgrowth*step*v_set_timestep) # diameter increasing with time
variable cuttime equal v_diamtime*5
fix growth all adapt 1 atom diameter v_diamtime
pair_style granular ghost yes
pair_coeff * * hooke ${Kn} ${dissip} tangential linear_nohistory 1.0 ${muFrict} cutoff ${cuttime}
thermo ${thermo_num}
thermo_style custom step etotal
dump growthdmp all custom ${thermo_num} dumpgrowth.lammpstrj id type x y z radius
variable run_growth equal floor((v_growth_ratio)/(v_radgrowth*v_set_timestep))
run ${run_growth}
Have you tried not setting an explicit cutoff but forcing the neighbor list rebuild in every step with?
neigh_modify delay 0 every 1 check no
The neighbor list is built from the sum of the two radii plus neighbor list skin. With a large enough skin, you may be able to increase the “every” value and thus tune the computational cost (a larger skin increases it, an every 2 or every 3 will then cut it in half or to a third).
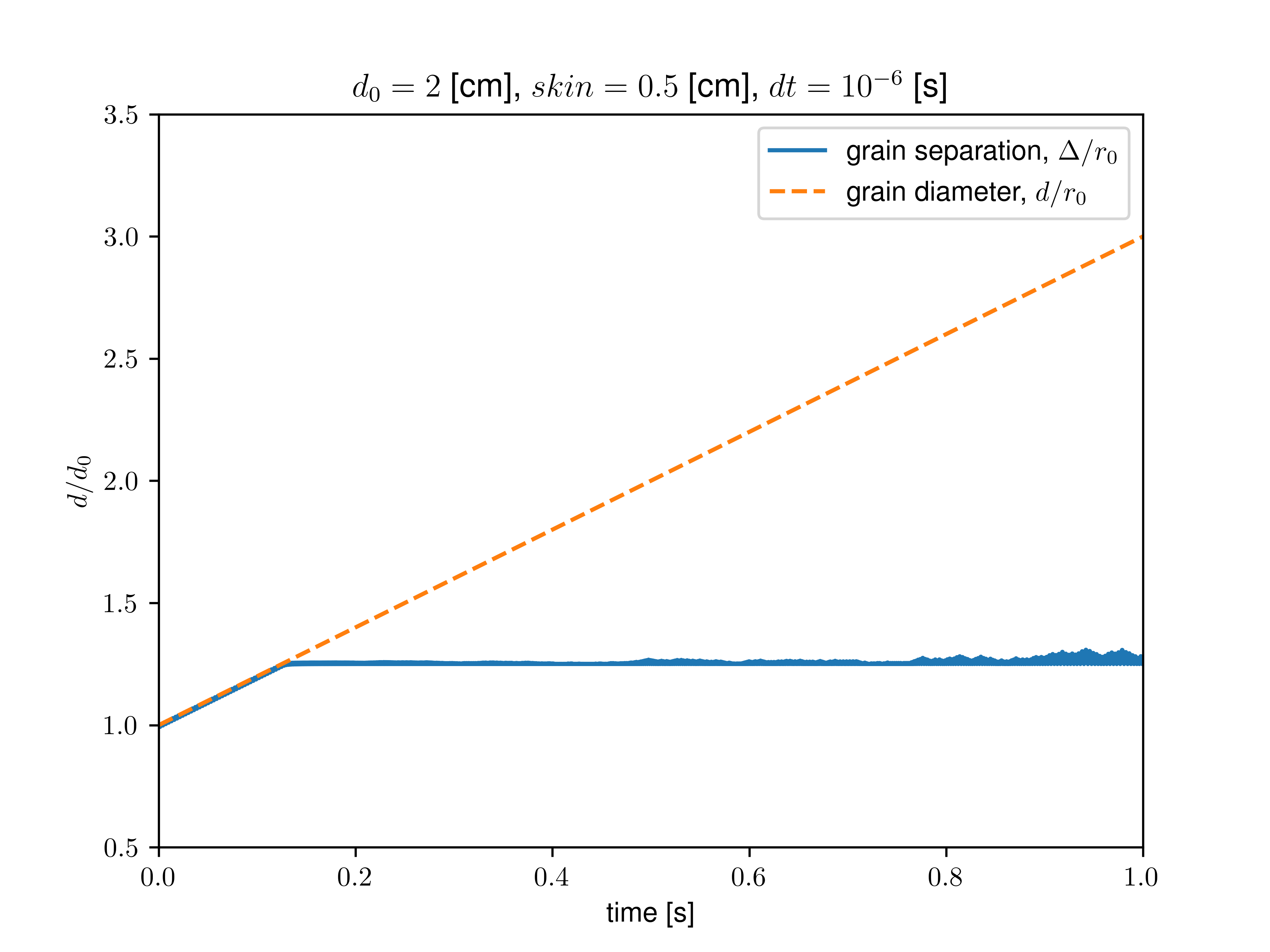
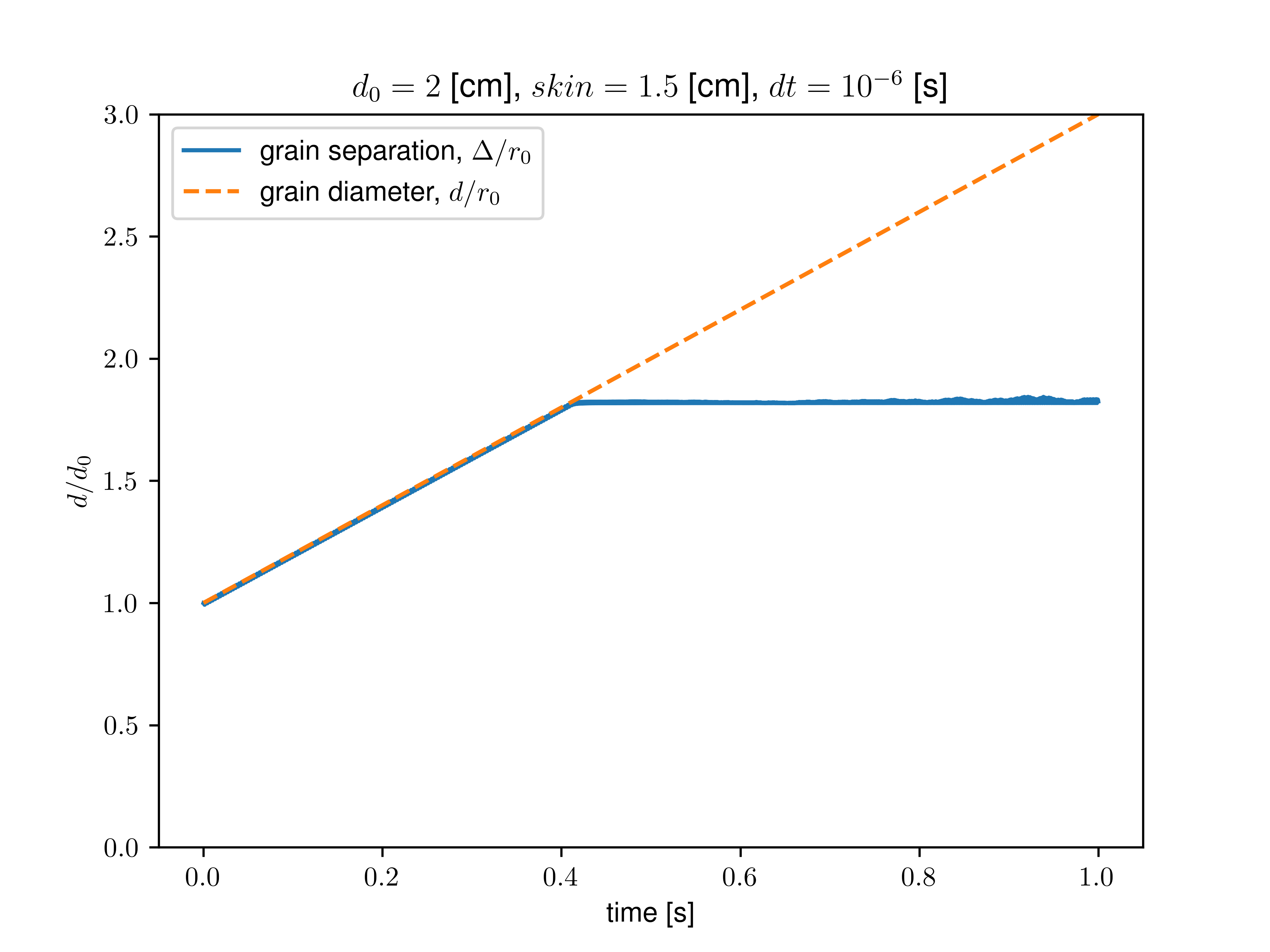
I played with the values of “neigh_modify every” and “neighbor skin” and removed the imposed cutoff as suggested. However, it seems that when rebuilding the neighbor list, only the initial values of the radii are taken into account, not the current ones modified by the fix adapt.
For instance, setting every 1 with a skin value equal to the initial radius, the grains start overlapping when their radius is about twice their initial one.
Nonetheless, when setting a large skin value (larger than the final size of the grown grains), they do not overlap, and the computation time is improved by increasing the “every” value.
Sorry, but that is not supported by the source code. The system does not store those initial radii. Only fix adapt keeps a copy if so instructed in order to be able to reset those values.
Don’t you get a warning that atoms are time integrated multiple times with this?
Using those two time-integrating fixes together for the same atoms is definitely an error and can lead to bogus trajectories.
Oh, I see. Then I am lost as to why rebuilding the neighbor list at each step fails to detect the neighbors after some growth.
I was indeed not sure if that was permitted, thank you for the clarification. It didn’t produce a warning (LAMMPS version 20230802), and removing the second fix did not change the outcome of the computation.
I’ve experimented with your input a little bit and it seem to me that you are misinterpreting the results.
The situation changes if I comment out fix viscous or make the applied friction much smaller. My interpretation of the results you see is that you are draining any kinetic energy from the system faster than it can create it from the repulsion and thus at some point the atoms freeze and cannot react to the repulsive force.
The warning must be there. The corresponding code and flags have not changed for either of those two fixes in more than 15 years.
Thank you very much for your time and insights; I really appreciate it.
I indeed tried to dissipate kinetic energy as quickly as possible to keep the grains in close contact and prevent them from drifting too far apart (my application requires to be in an overdamped regime).
For instance, when I comment out the fix viscous, the initial contact will push them apart, and they will continue drifting apart faster than their radius growth.
If my grains stop pushing each other because I am removing kinetic energy too fast, would we expect changing the skin value to affect the size at which the grains stop moving?
Leaving all other parameters unchanged, this critical size seems proportional to the skin value. Wouldn’t that indicate that there is also a neighbor detection issue?
My apologies the warning was indeed there. WARNING: One or more atoms are time integrated more than once (../modify.cpp:289), I thought that it would cause an error preventing the code from running.
If you remove too much kinetic energy, then the atoms will overlap too much because of the growth of the radius being faster than they can react to it and move apart and then eventually you will have invalid forces from pair style granular since the atoms are in an unphysical state. To handle this situation correctly, you would have to use a correspondingly shorter timestep value. Please also keep in mind that with the growth of the diameter, the mass of the atoms grows (the density remains constant by default and you didn’t tell fix adapt to keep the mass unchanged) and thus the atoms’ inertia grows with increasing diameter.
You can monitor the displacement by adding this variable definition and using the custom thermo style
variable dely equal sqrt((y[1]-y[2])*(y[1]-y[2]))
thermo_style custom step ke v_dely fmax
If you keep the friction at 0.001, even a timestep of 0.000001 seems to be too large as the displacement slows eventually down.
If you keep the timestep and reduce the friction to 0.0001, the dynamic balance seems to be retained. Even though it is “on the edge” the force remains for the most part constant, i.e. you have a dynamic equilibrium between the growth of the diameter and the pushing of the atoms apart.
Thank you very much for your precious explanations. I am very new to molecular dynamics, so I underestimated the critical roles of inertia and timestep values. I will estimate the various timescales involved in this system more carefully to make it behave correctly.
Again, thank you very much for your time and support.
As an aside, the command compute pair/local should be helpful in diagnosing the balance of forces. It’ll let you access forces between atoms to confirm the contact is correctly detected and compare the magnitude of different contributions using p1 through pn attributes (described at the bottom of the Mixing section in pair granular).
Thank you very much for the tip about this super helpful command!
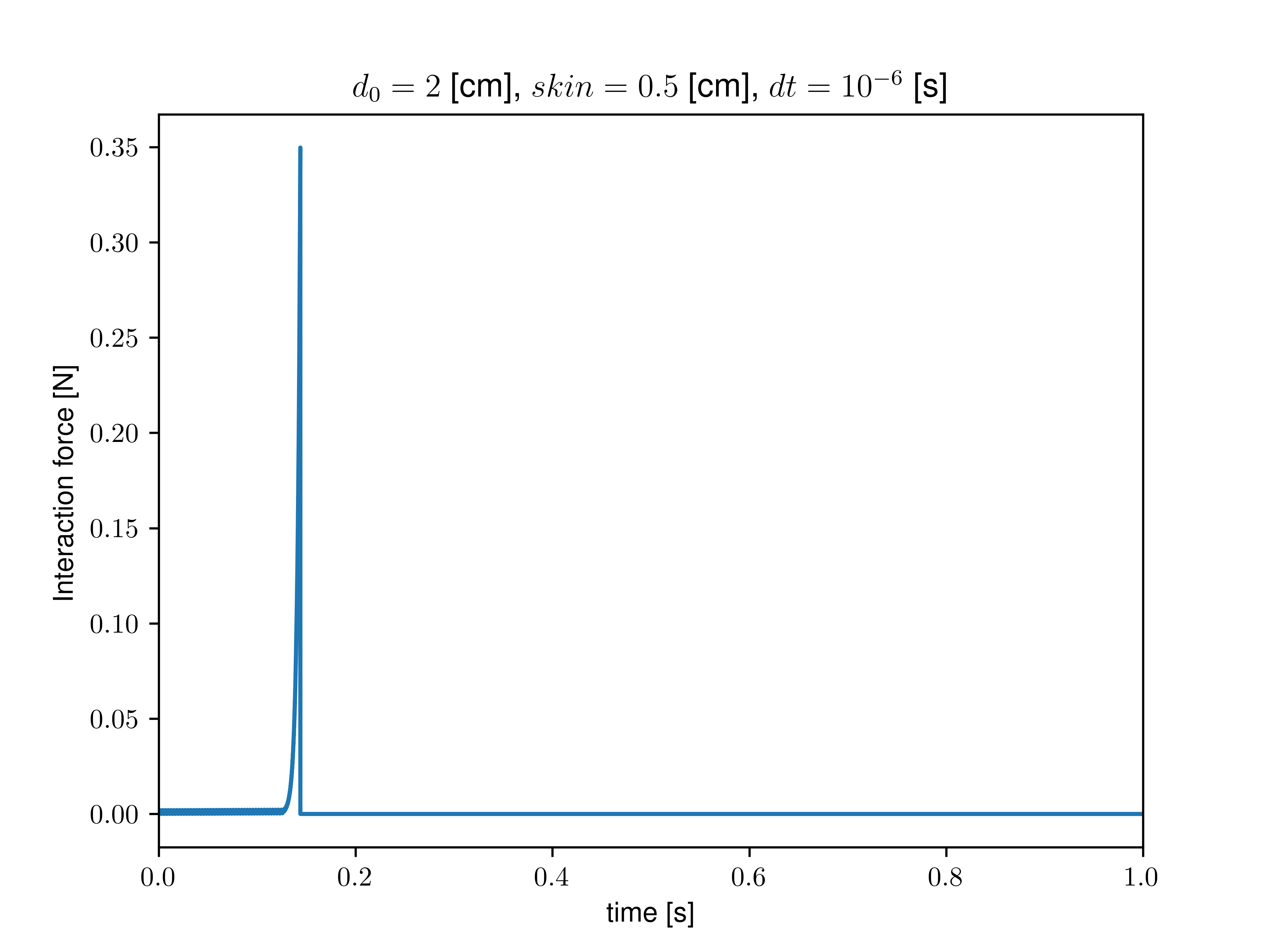
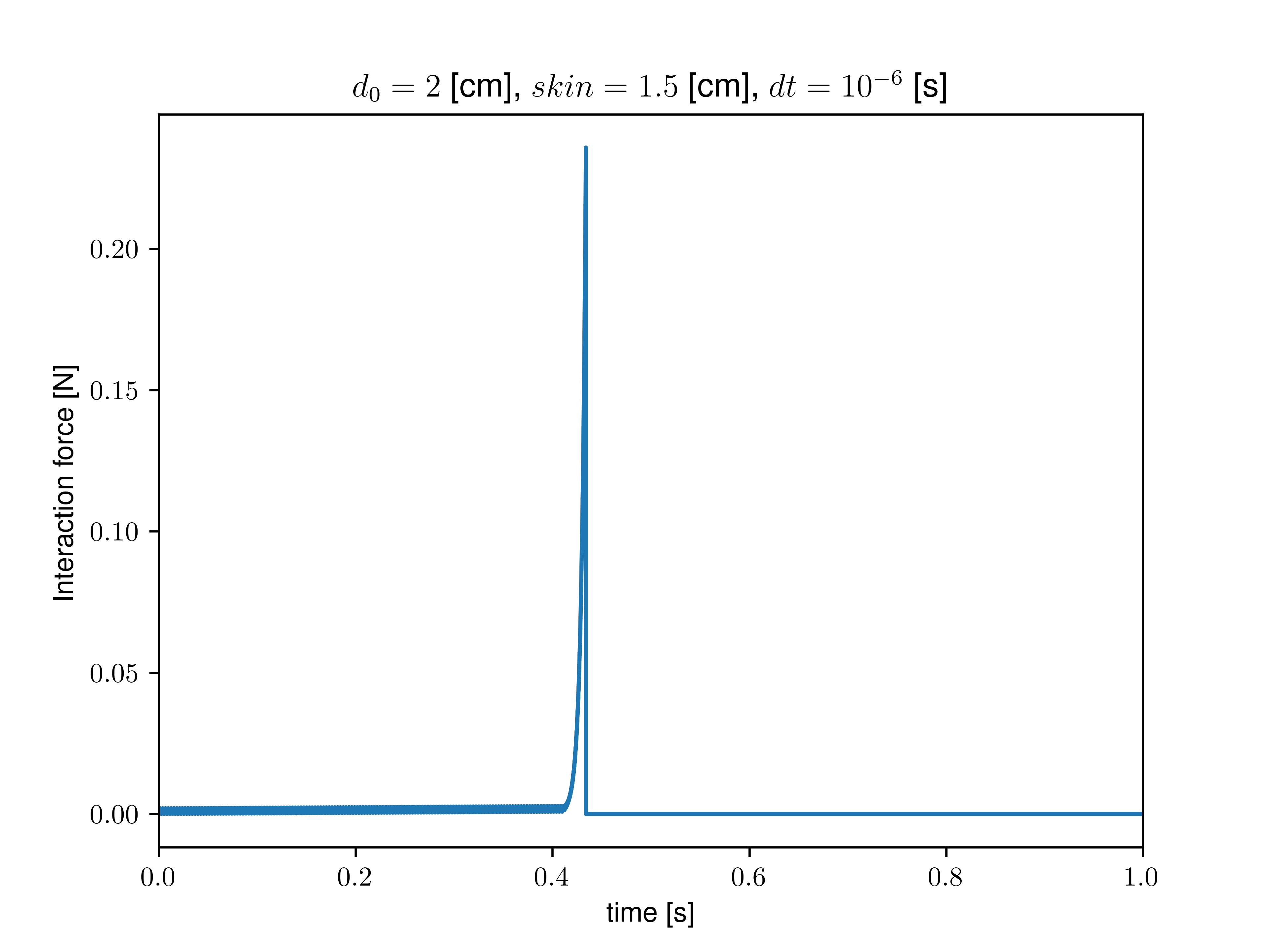
I then proceeded to measure the interaction force explicitly with compute pair/local and the neighbors with compute pair/local. I removed all sources of dissipation (no fix viscous and zero dissipation/friction in the interaction), kept mass constant during growth, and calculated the neighbor list at each step. I added a soft (1/100 of grain stiffness) confining spring force to avoid the grains flying away at the first contact. In that case, after some growth, I still find a loss of contact (interaction force goes to zero, no more neighbor detected). This critical growth size depends on the set skin value and seems independent of the timestep or confining stiffness.
As we get closer to the critical growth size, we indeed observe a divergence of interaction forces (see graph below). However, as I removed all apparent dissipative forces, I am still confused about where this limiting timescale (lengthscale) may emerge.
Another thing to keep in mind is while the neighborlist uses the current radii to determine whether two particles are neighbors (as Axel noted), the radius of the binlist stencil is based on the initial estimates of pair cutoffs from initial atom radii. If those initial estimates are too small, the stencil cannot be guaranteed to include all neighbors.
If you want to override this, pair granular has an optional cutoff option. The doc page discusses it in the context of fix adapt. Maybe this is what you need?
Oh, that’s very enlightening on how neighbors are constructed. Thank you very much.
The doc page indeed mentions that the default radius-based cutoff may not be adequate when the radius is increasing, for instance. However, this cutoff value is set once when defining the interaction and cannot be modified over time with a fix_adapt. Setting a large enough value initially takes huge computing time at the beginning if we start with many small particles.
I will try to divide the growth into multiple shorter runs, redefining the cutoff before each run. That way, I should be able to keep the actual grain size and cutoff values compatible during each run.
Thank you again, everyone, for the help; I really learned a lot.
is probably not true. The documentation for pair granular specifies that by default there is viscoelastic damping with a damping coefficient that increases with grain size. I would not be surprised if you have two beads that are simply being damped into place as their size increases unreasonably.
is a premature worry. You should start by experimenting with a much larger cutoff and verify, not just suspect, that it will lead to significant neighbour list construction slowdown.
Don’t forget that neighbour list checking and building intervals are much easier to customise mid-run. A large neighbour search radius will also result in neighbour lists that “expire” less quickly, so (I think) you can also adjust the neighbor keywords every and delay to save some time.
Thank you very much for your comments. Regarding the viscoelastic damping in the pair granular, I set the corresponding damping coefficient to zero, so I thought it would eliminate this dissipating component. However, the sharp force increase before the contact loss seems to indicate that there is still a dissipative element somewhere.
I actually started with a large number of grains. Faced with computing times much larger than expected for such simulations, I decided to focus on the two-grain system to understand why I needed such a large cutoff value. Thanks to the various replies, I now understand better how neighbor lists are built and how to customize that part, which will help me reduce computation time in a controlled way.