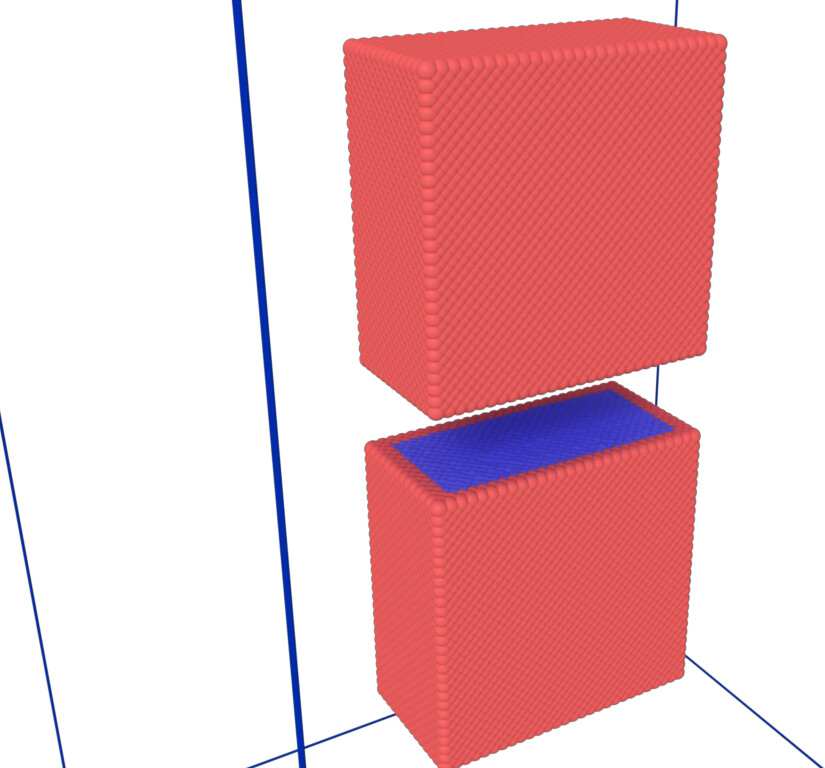
Hi !
I am a first-year graduate student, recently began to use LAMMPS application in EDM analysis, the specific effect I want to do and my code is as follows, please help me to see what the problem is.
Initialization
units metal
dimension 3
boundary p p p
atom_style atomic
#Define the simulation area
region whole block 0 300 0 300 0 500 units box
create_box 2 whole
Create an electrode
lattice fcc 3.615
region electrode block 100 200 100 150 230 330 units box
create_atoms 1 region electrode
group electrode region electrode
Create artifacts
lattice fcc 3.615
region workpiece block 100 200 100 150 100 200 units box
create_atoms 2 region workpiece
group workpiece region workpiece
Define the side boundary layer area of the electrode
region el_side2 block 195 200 100 150 230 325 units box
region el_side3 block 105 195 100 105 230 325 units box
region el_side4 block 105 195 145 150 230 325 units box
region el_side5 block 100 200 100 150 325 330 units box
region wp_side1 block 100 105 100 150 100 200 units box
region wp_side2 block 195 200 100 150 100 200 units box
region wp_side3 block 105 195 100 105 100 200 units box
region wp_side4 block 105 195 145 150 100 200 units box
group el_side2 region el_side2
group el_side3 region el_side3
group el_side4 region el_side4
group el_side5 region el_side5
group wp_side1 region wp_side1
group wp_side2 region wp_side2
group wp_side3 region wp_side3
group wp_side4 region wp_side4
The thermostatic region of the electrode
region el_hwc block 105 195 105 145 305 325 units box
region wp_hwc block 105 195 105 145 100 120 units box
region hwc union 2 el_hwc wp_hwc
group el_hwc region el_hwc
group wp_hwc region wp_hwc
group hwc region hwc
Newtonian layer
region el_ndc block 105 195 105 145 230 305 units box
region wp_ndc block 105 195 105 145 180 200 units box
region ndc union 2 el_ndc wp_ndc
group wp_ndc region wp_ndc
group el_ndc region el_ndc
group ndc region ndc
Define atomic type and mass
mass 1 63.546 # Cu
mass 2 63.546 # Cu
Select the applicable force field
pair_style eam/alloy
pair_coeff * * Cu_Zhou04.eam.alloy Cu Cu
Fixed the motion of atoms in the boundary layer on four sides
fix fix_el_side2 el_side2 setforce 0.0 0.0 0.0
fix fix_el_side4 el_side4 setforce 0.0 0.0 0.0
fix fix_el_side5 el_side5 setforce 0.0 0.0 0.0
fix fix_wp_side1 wp_side1 setforce 0.0 0.0 0.0
fix fix_wp_side2 wp_side2 setforce 0.0 0.0 0.0
fix fix_wp_side4 wp_side4 setforce 0.0 0.0 0.0
#Set the initial temperature
velocity all create 300 12345
Application of constant temperature layer
fix nvt_hwc hwc nvt temp 300 300 0.1
Workpiece temperature
fix 2 wp_ndc nvt temp 300 300 0.1
Apply temperature control to the electrode
fix 3 el_ndc nvt temp 300 300 0.1
A variable that controls whether the heat source is activated or not, stop heating after 15000 steps
variable heat_active equal “step < 15000”
variable heat_mult equal “v_heat_active * 20.8”
variable heat_mult_2 equal “v_heat_active * 8.8”
The first heat source
variable x0 equal 120
variable y0 equal 115
variable z0 equal 240 # Central position of heat source
variable Pm equal ${heat_mult} # Maximum heating intensity
variable k equal 0.03 # Parameters that control the range of Gaussian distribution
variable fx atom v_Pmexp(-v_k((x-v_x0)^2+(y-v_y0)^2+(z-v_z0)^2))
variable fy atom v_Pmexp(-v_k((x-v_x0)^2+(y-v_y0)^2+(z-v_z0)^2))
variable fz atom v_Pmexp(-v_k((x-v_x0)^2+(y-v_y0)^2+(z-v_z0)^2))
Applying the force of Gaussian distribution to the Newtonian layer
fix heat el_ndc addforce v_fx v_fy v_fz
The second heat source
variable x0_2 equal 155
variable y0_2 equal 130
variable z0_2 equal 210
variable Pm2 equal ${heat_mult_2}
variable k equal 0.03
variable fx_2 atom v_Pm2exp(-v_k((x-v_x0_2)^2+(y-v_y0_2)^2+(z-v_z0_2)^2))
variable fy_2 atom v_Pm2exp(-v_k((x-v_x0_2)^2+(y-v_y0_2)^2+(z-v_z0_2)^2))
variable fz_2 atom v_Pm2exp(-v_k((x-v_x0_2)^2+(y-v_y0_2)^2+(z-v_z0_2)^2))
fix heat2 wp_ndc addforce v_fx_2 v_fy_2 v_fz_2
thermo 500
thermo_style custom step temp pe ke etotal press pxx pyy pzz
dump 1 all custom 500 dump.Cu.xyz id type x y z
run 60000
I follow the production of this article, I feel that there is something wrong with my Gaussian heat source setting, because this article does not specify how to set it.(Influence of discharge gap on material removal and melt pool movement in EDM discharge process (https://doi.org/10.1007/s00170-021-08577-z))
I think the key lies in the setting of the heat source. I am not sure how to set the heat source properly to achieve the effect in the article. Of course, my other settings are also rough. Please give suggestions.