I cannot really help with VACF command because i haven’t used it.
I usually produce velocity vectors using Lammps and do post-processing with those velocity vectors using Matlab because it helps me to do more careful signal processing with some details. If your system is well-equilibrated and produce good velocity vectors, then you can try doing the postprocessing without using VACF command.
Is fix ave/time definitely returning the output you want? At the moment, fix ave/time is going to give you an output on every 1000th timestep, where the value returned is an average over the previous 100 values of the VACF at 10 timestep intervals. You are smoothing out some of the time dependence. It would be more typical to just sample this every timestep.
Thanks,
Niall
Niall Jackson
PhD Student, Bresme Group (Computational Chemical Physics Section)
Department of Chemistry, Imperial College London
Thanks for you reply. Actually I have tried to vary the number of time steps to take from 1,10,100 and 1000 but didnt get better and dont know where things went wrong.
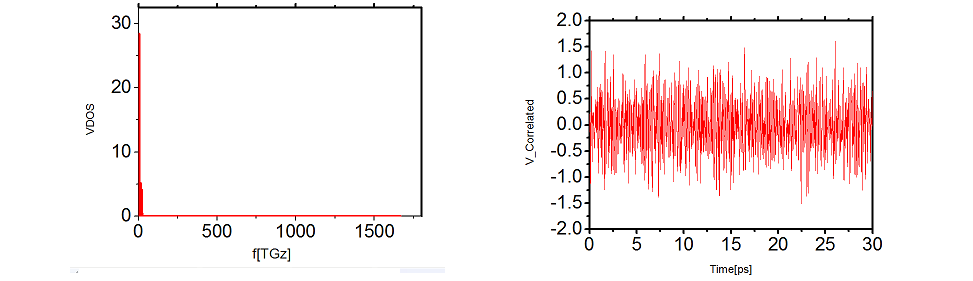
I have attached how the PDOS(Fist figure) and the correlated (Second figure)data looks like taken by setting " ave/time 1 1 1 c_10[4] ".
That VACF looks very noisy - how many atoms are in the group sub2?
I should point out that compute VACF isn’t doing every possible correlation. For each atom, it correlates the velocity on the current timestep with v(0), the velocity at the start of the run. This means that your statistics in <v(0)v(1)>, for example, don’t increase with the length of the simulation. It works well if you have a large number of atoms in the group, though.
if somebody here has interest in a C++ programming project, there is a
way to simplify the whole process and take out some of the arbitrary
choices, by assembling the spectral densities directly in fourier
space. the downside is increased use of RAM for the intermediately
accumulated vector data. it would work not only for individual
velocities, but also on per-molecule/chunk dipole moments or the total
dipole moment of the system (that is how it was originally conceived
in combination with ab initio MD simulations).
the functionality is currently available in VMD and could be used in
principle with existing LAMMPS trajectory dumps, either by using
coordinates and reading partial charges from the data file, or the
atomic velocities if they are included, however, the space
requirements in the latter case for a decent resolution from classical
MD are huge, so that a direct integration into LAMMPS would be
desirable. even in the former case, it might be desirable to compute
the total dipole in every step and write it out to a particular file
and then read that file into VMD as a long list of vectors and then
compute the spectral density. please note that this plugin is
basically a standalone program (there is a fortran version of it
bundled with CPMD) and does not use any VMD internal features.
i've been considering repeatedly to port this over, but this is really
a perfect project for somebody to learn about integrating code into
LAMMPS. i'd be willing to provide assistance for as long as doesn't
become more work than doing it myself.