Hello,
I am trying to simulate C60 buckyball impact on graphene sheet. When i run my script its getting stuck after defining lattice parameters. I have attached the relaxation script below. Kindly help
Hello,
I am trying to simulate C60 buckyball impact on graphene sheet. When i run my script its getting stuck after defining lattice parameters. I have attached the relaxation script below. Kindly help
Hello,
I am trying to simulate C60 buckyball impact on graphene sheet. When i run my script its getting stuck after defining lattice parameters. I have attached the relaxation script below. Kindly help
this is due to the *EXTREMELY* inefficient way how you define your
simulation cell: you define a *GIGANTIC* (periodic) cell (which will
lead to *EXTREMELY* inefficient parallelization) for no good reason.
you can achieve the same effect much better by simply not using
periodic boundaries (and without having to fear losing atoms) with
"boundary m m m" and then you can define your simulation cell to just
barely contain your created atoms, e.g. as:
region box block -1 16 -1 16 -1 26
the reason, that LAMMPS gets stuck is because it iterates over all
grid points in the *ENTIRE* box (which are of the order of 10**20) to
fill two comparatively *TINY* regions. this nested loop will take a
very, very long time.
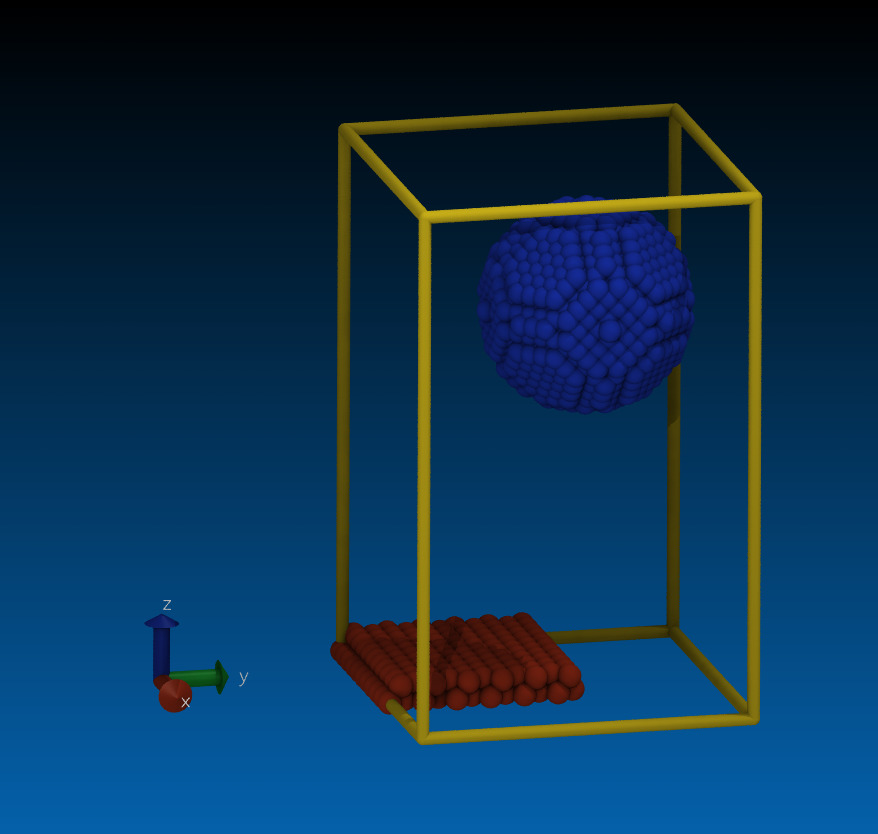
that said, your create_atoms commands create neither a graphene sheet
not a bucky ball as evidenced by the attached snapshop.
Does the “lattice fcc 1 spacing 1.42 1.42 1.42” command with “region region7 sphere 10 10 20 5 units box” create C60 buckyball?
C60 buckyballs has 60 carbon atoms. Similarly, C180 has 180 carbon atoms.
In one of our papers, we studied C180 buckyball impact on graphene sheet. Hope this paper helps.
https://www.sciencedirect.com/science/article/pii/S0008622316301269
Sanjib
Can you please suggest how i can simulate graphene and buckyball. I used hex lattice for graphene but lammps is giving me error since its a 3d simulation and hex is a 2D lattice.
Can you please suggest how i can simulate graphene and buckyball. I used hex lattice for graphene but lammps is giving me error since its a 3d simulation and hex is a 2D lattice.
yes, the manual clearly states that, but you may need to update your
crystallography knowledge. what does "hcp" stand for?
as for getting graphene and/or buckyball structures. you can start
here: http://bfy.tw/LgLN
axel.
Thankyou for your time Dr Kohlmeyer
I started using lammps just about two weeks and didn’t not go through the documentation thoroughly. This is the reason I’m very vague in what I’m doing. I have really limited time to submit my project so I didn’t pay proper attention to the docs. I realised my mistake now.
Thanks for guiding me in generating graphene structure.
Thankyou for your time Dr Kohlmeyer
I started using lammps just about two weeks and didn't not go through the documentation thoroughly. This is the reason I'm very vague in what I'm doing. I have really limited time to submit my project so I didn't pay proper attention to the docs. I realised my mistake now.
you will need more input than just the documentation. your input deck
is full of examples showing, that you don't understand what you are
doing. understanding is not something that you can obtain in a couple
of weeks and most certainly not from a software manual.
i always keep telling people that a software manual is like the manual
you get with a car: it tells you where all the knobs and switches are
and what features are available and how to use them, *but* it does not
teach you how to drive a car and how to follow the rules and laws (of
computers and physics in the case of software).
thus it is highly unlikely, that you'll be able to build a meaningful
and correctly working simulation input unless you obtain some proper
and competent local and in-person tutoring. a mailing list cannot help
you there.
axel.
Thankyou Dr Kohlmeyer
I’ll keep that in mind. Thanks for the support.
I have generated an input file but the following error is occurring
reading atoms …
ERROR on proc 0: Invalid atom type in Atoms section of data file (…/atom_vec_atomic.cpp:617)
Last command: read_data ${input}
#------------input file----------------------
1660 atoms
2 atom types
-150.00 150.00 xlo xhi
-150.00 150.00 ylo yhi
-300.00 50.00 zlo zhi
Masses
1 12.01100000
2 12.01100000
Atoms
1 1 -0.00000000 2.46000000 1.70000000
2 1 0.71010000 1.23000000 1.70000000
3 1 2.13040000 1.23000000 1.70000000
4 1 2.84060000 49.20000000 1.70000000
5 1 -0.00000000 39.36000000 1.70000000
6 1 80.94000000 41.82000000 1.70000000
7 1 83.07040000 40.59000000 1.70000000
8 1 -0.00000000 41.82000000 1.70000000
9 1 0.71010000 40.59000000 1.70000000
10 1 2.13040000 40.59000000 1.70000000
11 1 4.26000000 41.82000000 1.70000000
12 1 76.68000000 44.28000000 1.70000000
.
.
.
.
.
1638 2 -0.69230000 4.21310000 3.92070000
1639 2 -2.30450000 -0.74880000 3.92070000
1640 2 -2.75690000 0.64350000 3.92070000
1641 2 1.91630000 -1.80050000 5.16600000
1642 2 3.03650000 -0.98660000 5.16600000
1643 2 -1.57260000 -0.98660000 5.16600000
1644 2 -0.45240000 -1.80050000 5.16600000
1645 2 -2.30450000 1.26610000 5.16600000
1646 2 -1.87670000 2.58300000 5.16600000
1647 2 0.03960000 3.97530000 5.16600000
1648 2 1.42430000 3.97530000 5.16600000
1649 2 3.76850000 1.26610000 5.16600000
1650 2 3.34050000 2.58300000 5.16600000
1651 2 0.73200000 -1.41570000 5.93560000
1652 2 -1.57260000 0.25860000 5.93560000
1653 2 -0.69230000 2.96780000 5.93560000
1654 2 2.15620000 2.96780000 5.93560000
1655 2 3.03650000 0.25860000 5.93560000
1656 2 0.00000000 2.01490000 6.66360000
1657 2 1.46390000 2.01490000 6.66360000
1658 2 1.91630000 0.62260000 6.66360000
1659 2 0.73200000 -0.23780000 6.66360000
1660 2 -0.45240000 0.62260000 6.66360000
I have generated an input file but the following error is occurring
reading atoms ...
ERROR on proc 0: Invalid atom type in Atoms section of data file (../atom_vec_atomic.cpp:617)
check your data file! it is likely not correctly formatted or has
incorrect content (atom type set to be smaller than 1 or larger than
2). the error message is pretty self explanatory and nobody here has
mind reading abilities to tell which error you made exactly. remember
what i told you about "driving school" and "finding a tutor". this is
a question you should not be posting to this mailing list unless you
have *positive* proof, that you have found a bug.
if you do things correctly, it will work, and since you are using
functionality that is *very* widely used, it is *extremely* unlikely,
that you are running across a yet undiscovered bug. and much more
likely that you made a mistake. figuring those out and solving them is
part of learning how to be a scientist. this mailing list is not the
place to learn it.
i am generally supportive of beginners, but that reaches only so far,
and at some point it is crucial, that you learn how to resolve
(trivial and self-evident) issues yourself or else i will simply
filter any of your e-mails out, when i get the impression that you are
not putting in the kind of effort and thinking required yourself and
are depending too much on people like me to solve problems that you
should be able to solve yourself, if you want to succeed at doing MD
simulations.
axel.