I’ve been utilizing LAMMPS for my simulations involving yarn structures, where I model the yarns as particles connected by harmonic potentials to represent bonds and angles. Additionally, I’ve used soft pairwise interactions to simulate the collisions between these particles. So far, this approach has yielded satisfactory results.
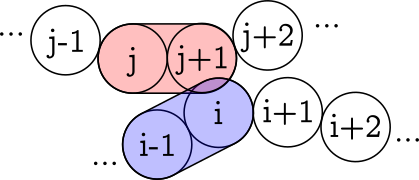
However, I’m now interested in exploring a new direction for my simulations. Specifically, I’d like to shift from modeling the interactions between particles as if they were spherical to treating them as if they were capsules. This means envisioning each yarn segment as a capsule, constituted by pairs of bonded particles. Therefore, a collision between two yarns would involve the interactions between four particles (2 from each yarn).
I didn’t find anything in this direction in LAMMPS (maybe there’s a reason that I’m missing). I wonder if anyone in the community has attempted something similar or could offer guidance on how to implement this capsule-like pairwise interaction model in LAMMPS. Any advice, references, or starting points would be greatly appreciated.
Here is a 2008 paper on Monte Carlo simulation of bowl-shaped particles. It should be possible to implement that pair function leveraging the Gay-Berne and/or the RE-squared potentials.
@silvap1 please keep in mind that people here are not familiar with your research and thus have no idea what a “yarn” structure would be, how you exactly applied which potentials, what you understand under a “capsule-like” structure and how that differs from what you did before or what specific functionality you would need.
So when you are asking for assistance you will first have to explain these things, e.g. by showing some graphics and some simple and small yet representative input deck for a “yarn” system and some graphics in how you would see a “capsule” structure.
Since most of the functionality in LAMMPS is focused on molecular systems, it is not a surprise that you cannot find what you are looking unless you would be using terms that would be consistent of such systems (rather than what is common in your research).
For example for the term
You can easily have this, but from your description it is not clear which bonds go from where to where.
I am currently checking the approach suggested by f@hothello . The suggestion would require (if I understood correctly) changing particles to ellipsoid-type ones. I am trying to figure out if the alignment of the ellipses would match with the alignment of the bonds or if somehow they become misaligned and strategies to overcome it.
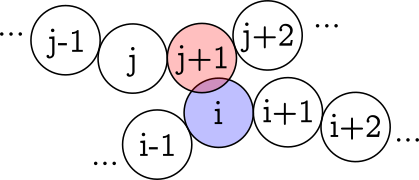
I am sorry for not being clear enough. My yarns start from a bead-spring model where the radius of the bonds are equal or lower twice of the radius of the beads. The bonds link consecutive beads (…, i-1 bonded with(<->) i, i ↔ i+1, i+1 ↔ i+2, …):
I also include a bending rigidity term by considering the angle made by 3 consecutive beads. And then I set the potentials as:
Currently, to avoid yarns from passing through each other, I set a soft potential:
pair_style soft 1.0
pair_coeff * * 100.0
Everything works as expected. Now, even not setting any potential for simulating friction between pairs, the spherical topology makes yarns slide through “dents” between the spheres. I was hoping to find a strategy that could somehow fill those gaps. I thought that if somehow instead of calculating the distance between two particles, I calculate the distance between two capsules (made by 2 consecutive particles)
I could calculate the repulsion force in a similar way to the soft pairstyle potential, with the fact that now that the contact will repel 2 particles at once instead of the typical 1 particle.
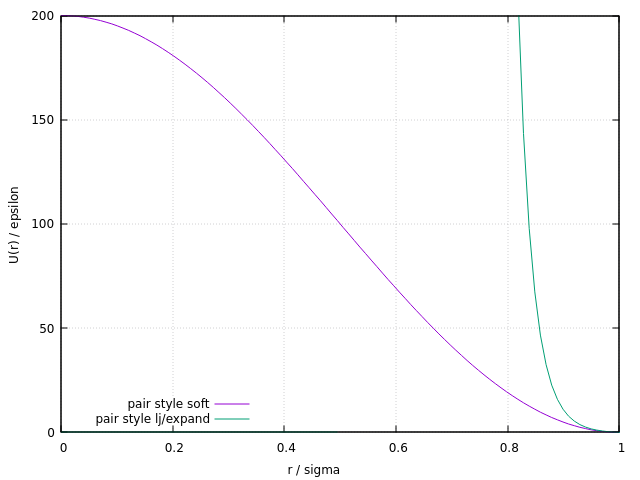
I would say the issue is more the softness and shape of your potential. If you want excluded volume, you need to use a potential that has a stiffer repulsion and some excluded “inner volume”. In the long run, you probably want to use pair style granular, but as an incremental change you could look at, for example, the lj/expand pair style. Below is a plot with the settings \epsilon = 0.25, \sigma = 0.5, \Delta = 0.5, r_c = 1.0 comparing that radial potential with your soft pair style settings (assuming reduced units).
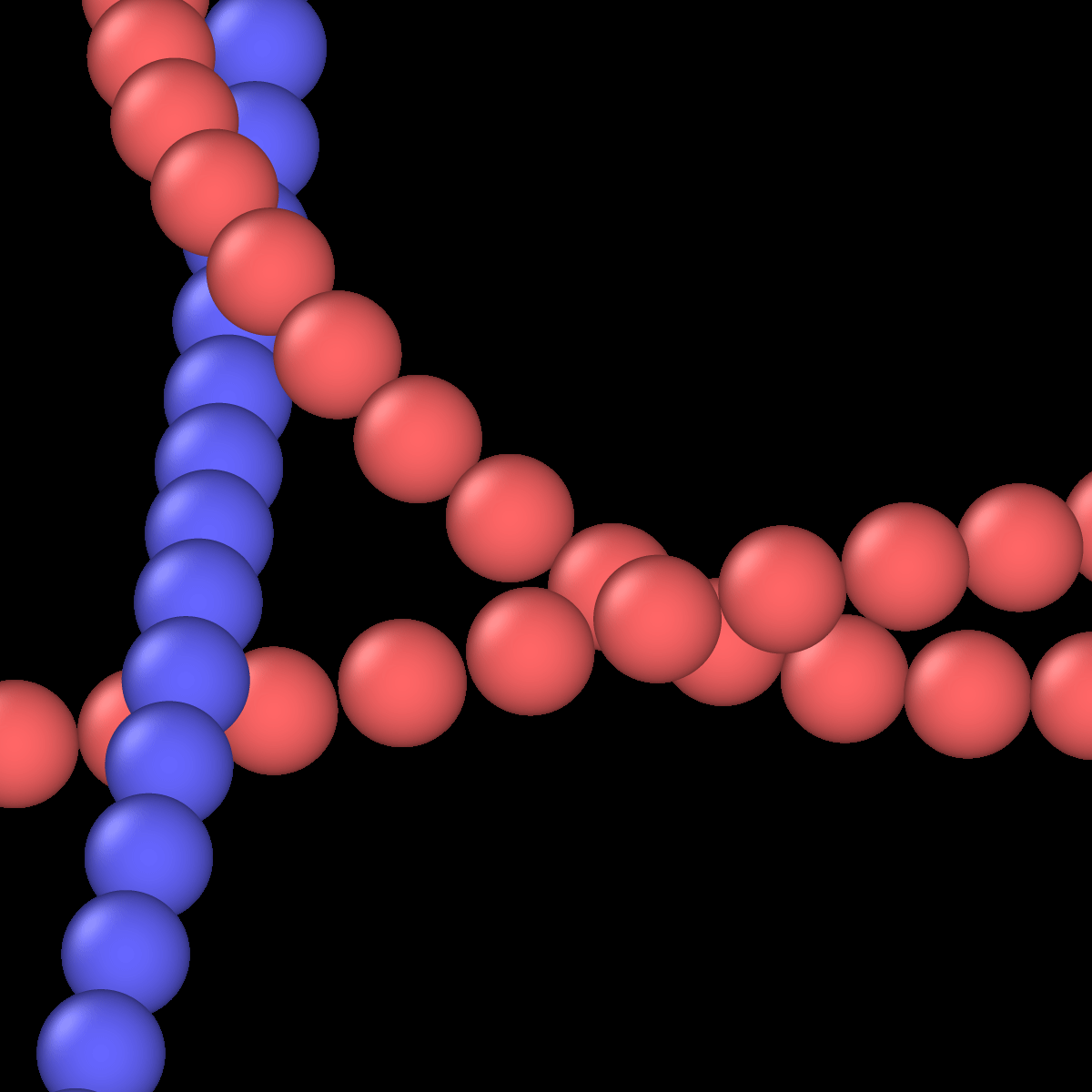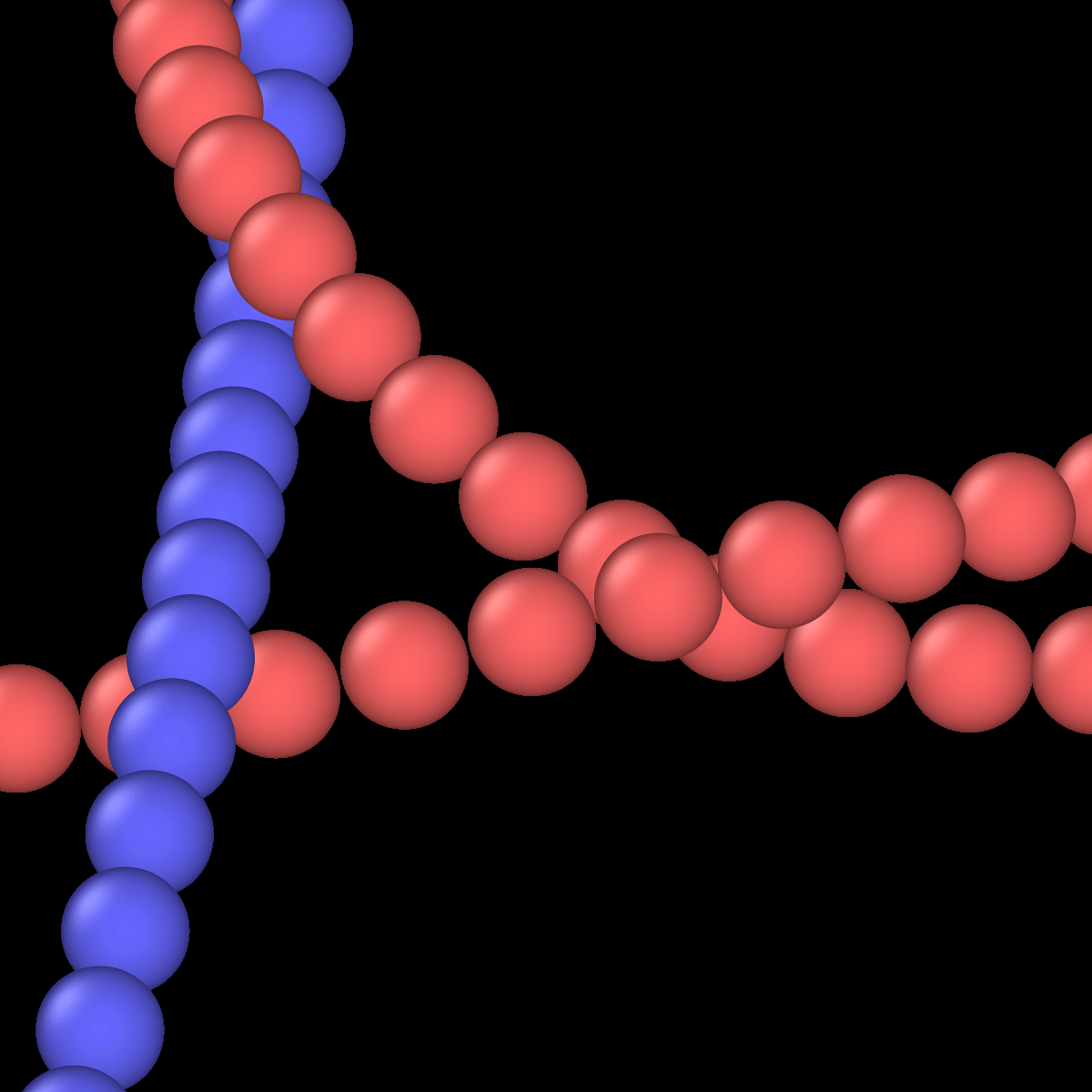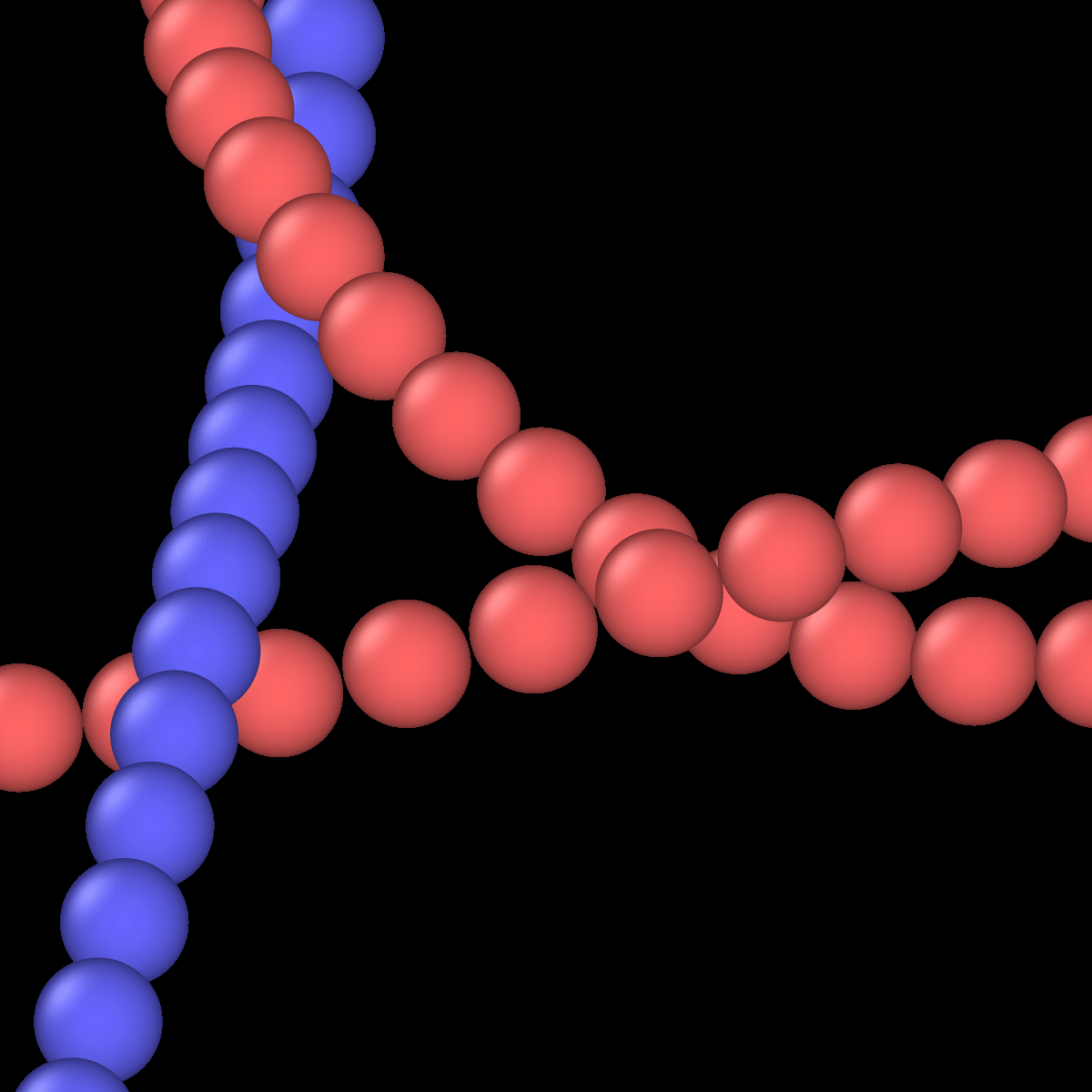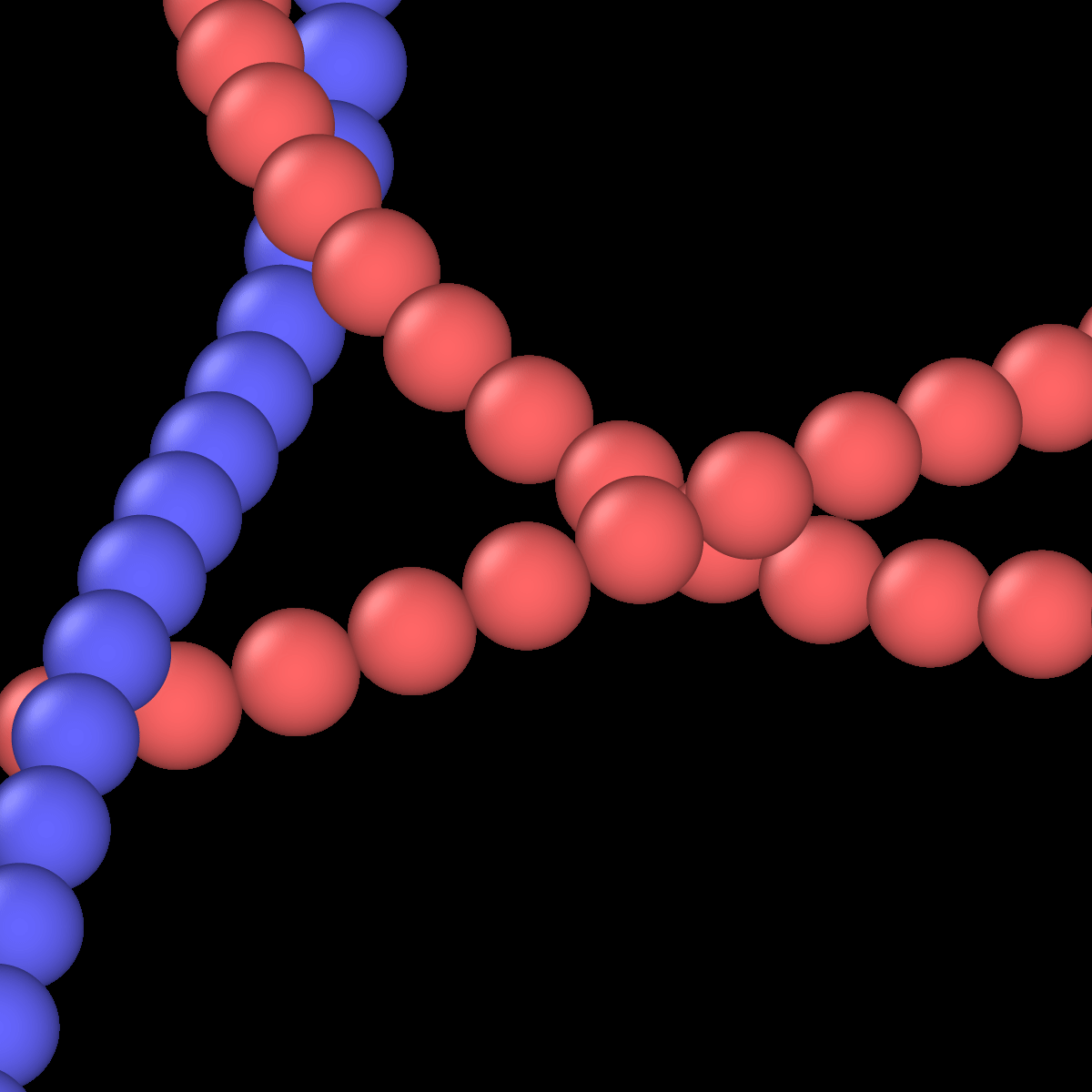
I changed to the stiffer potential you suggested. I still observe a similar behavior, which can be visually understood from the following sequence. When moving yarns “try” to move along other yarns, they are locked until building enough energy to jump to another gap:
I can increase the number of particles to mitigate part of the problem, which will reduce the size of the gap between spheres. However, this will also increase the number of neighbors and overall particles and consequently increase the computation times.
You have three parameters that you can experiment with: epsilon, sigma, and delta. Increasing delta will increase the inner volume, i.e. shift the repulsive branch of the potential to larger r, epsilon and sigma determine how steep the potential is and how far it extends beyond the excluded core. Technically speaking, to have a completely repulsive potential, you should have the cutoff set at 1.122462*sigma+delta. See the documentation for the pair style for details.
What is your special_bonds setting? for as long as the setting for bonds is 0.0 (the default), you can have overlapping beads, since the nonbonded interaction between the two atoms forming a bond is excluded and thus you won’t get an unphysical repulsion from having those beads overlap.
I understand I can change the shape of the potential. However, the problem seems to be how the potential works spatially. In simple terms, I would need the potential to consider all the space along the bond, not simply around the particle.
I am setting special_bonds, so I am using the default one.
As far as I know, there is no generic, “tube-like” potential in LAMMPS, but if you increase delta, there will be overlap between the beads and the larger this gets relative to the bond length to more the spheres will overlap and approximate a tube-like shape.
That is currently my strategy (but instead of increasing delta, I increase the number of particles which will result in something equivalent), however, wanted to reduce the number of particles to decrease the computation time, and wondered what would be my options and if it would be advantageous to develop a new fix (although I might be somehow limited to go that extent).
Thank you for your time
You are not using a particularly “expensive” potential and any kind of “shaped” interaction (like Gay-Berne ellipsoids) would be much more expensive to compute because of all the trigonometry required to get relative orientations.
In my personal opinion, your fastest path to results would be by trying to get access to a larger/faster/parallel computer. Many universities have such facilities and if that is not fast enough, you can apply for time at a national supercomputing center. Please note that pair style lj/expand is supported by both, the GPU package and the KOKKOS package for GPU acceleration. That can give you a significant speedup even on a local machine. For example am using an Intel NUC as desktop which has an Intel core i5-10210U mobile CPU with integrated GPU. This is supported by the GPU package when compiling for OpenCL and despite the modest CPU, I can get a significant acceleration from the GPU.
A quick update to this discussion. Increasing the number of particles will result, at some point, in neighbors particles to count towards pair wise interactions. The way I solved for my system was to exclude all intramolecular pair wise interactions by using the command:
neigh_modify exclude molecule/intra all
This works only because my system has no knots or the fibers/molecules don’t self-intersect ever. If there’s an alternative of excluding 1-5, 1-6, or even more pair wise interactions, I would like to learn about it.
If you are ok with adding particles (your original comments suggested not), then please have a look at pair style srp to avoid bond crossings: pair_style srp command — LAMMPS documentation