(It looks like my question never made it to mailing list. Reposting. Forgive me if you receive this message twice.)
Mahdi and Shyam and I have spent some time attempting to convert popular MSI force fields into moltemplate (.LT) format, including COMPASS, PCFF, CVFF, DREIDING, UFF etc. This is almost working.
But we are at an impasse and need help to make this available.
MSI .FRC files have a complicated atom (auto) equivalence system for generating bonded interactions. I could be wrong, but my suspicion is both moltemplate and msi2lmp sometimes fail to locate force field parameters for this same reason. Questions about this have been raised in the past:
https://sourceforge.net/p/lammps/mailman/message/29578563/
It could also be that the .FRC files we have are incomplete. Either way, it would be nice to know why these errors occur in moltemplate and msi2lmp.
----- example -----
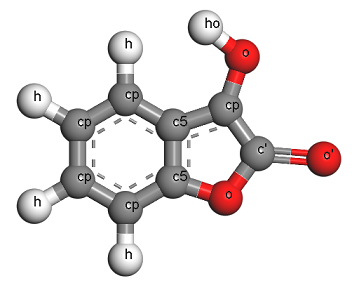
As an example, line 1117 of cvff.frc (in tools/msi2lmp/frc_files) defines parameters for a (3-body) class2 angle interaction between atoms of type:
cp o ho
The 3-body class2 angle interactions defined in “cvff.frc” contain bond-bond and bond-angle cross terms which depend on the equilibrium bond length between:
“cp” and “o”
“o” and “ho”
… however I could not locate a corresponding (2-body) bond interaction defined between atoms of type “cp” and “o” om the cvff.frc file. (I also had a chance to look at what I think is the commercial version of “cvff.frc” today, and I couldn’t find it there either.)
---- main question ----
Where are these mysterious bond lengths defined in FRC files? How should we look them up?
---- details (feel free to skip) ----
Relevant documentation on .FRC files can be found on page 402 of:
benedick.rutgers.edu/software-manuals/discovermanual.pdf
and pages 88, 90, 94, and 288 of:
http://benedick.rutgers.edu/software-manuals/forcefield.pdf
I confess that I hesitate to trust msi2lmp because it also fails to find the cp-o bond length, and it ignores the “auto_equivalences” section of the .FRC files.
I attached files for a molecule which contains this interaction “7511500.mdf”, “7511500.car”
Incidentally, for this example, we don’t have to worry about “equivalences”:
For those two atoms, “cp” and “o”, the same atom names are used consistently for looking up both mass, bond, and angle interactions. (In other words, the atom names do not change in the “equivalences” section of the “cvff.frc”. See lines 290 and 330 of that file.)
auto_equivalences?
Should we (also) be searching for the cp-o bond length by using the atom types from the “auto_equivalences” table? (“c_” or “cp_” and “o_”, in this example) Is that why we can’t find this bond-length?
When looking up bond-lengths used in 3-body (“bond-bond”) interactions, should we look up these bond-lengths using the atom names defined in the “bond” column of the “equivalences” table, …or the “angle” column? (they are note one-to-one.)
I’m curious how does EMC do it? (Is the source code for EMC available?)
Cheers
and thanks to anyone who finished reading this long post…
andrew
P.S. Even if we get this is working, there will still be serious benefits of using materials studio, such atom type assignment, and a graphical interface.
751150.mdf (1.76 KB)
751150.car (1.39 KB)
