Hello everyone,
I am trying to validate a paper on AlSi10Mg alloy:
“Effect of fabrication process parameters and graphene reinforcement on mechanical behaviour of additively manufactured AlSi10Mg alloy – A molecular dynamics simulation study.”
I am facing issues creating the correct hybrid potential for the alloy as described in the paper.
What I did so far:
- Built the atomic structure using Python, LAMMPS, and MATLAB
- After running the simulation, the atoms spread apart instead of forming a stable structure
- I suspect the issue is with my Lennard-Jones (LJ) parameters
- I cannot find suitable ε (epsilon) and σ (sigma) values for the cross-interactions:
Al–Si
Si–Mg
Al–Mg
Below is the input script I used (simplified):
LAMMPS input script for AlSi10Mg (incomplete example)
‘’’ units metal #gm/mole, angstrom, ps, ev, bars, K, gm/cc
dimension 3
boundary p p m
atom_style atomic
simulation box and lattice,potential files
lattice fcc 1.0
region whole block 0 60 0 60 0 60 units box
create_box 3 whole
read_data alsimg_sphere.data add append
-------------------- POTENTIAL SETUP --------------------
Atom type mapping:
1 = Al
2 = Si
3 = Mg
Hybrid/overlay: allows EAM, Tersoff, AIREBO, and LJ cross terms
pair_style hybrid/overlay eam/alloy eam/fs tersoff lj/cut 10.0
pair_coeff * * eam/alloy Al99.eam.alloy.txt Al NULL NULL
pair_coeff * * tersoff Si.tersoff NULL Si NULL
pair_coeff * * eam/fs Mg.eam.fs.txt NULL NULL Mg
Cross-interactions using Lennard-Jones
pair_coeff 1 2 lj/cut 0.01954 3.91728 # Al–Si
pair_coeff 1 3 lj/cut 0.01026 3.34978 # Al–Mg
pair_coeff 2 3 lj/cut 0.00915 3.25891 # Si–Mg
reset_timestep 0
timestep 0.0001
fix NVE all nve
thermo 100
dump mydump all custom 100 mydump*.txt id type x y z vx vy vz
run 1000
write_data mydata.data
quit ‘’’
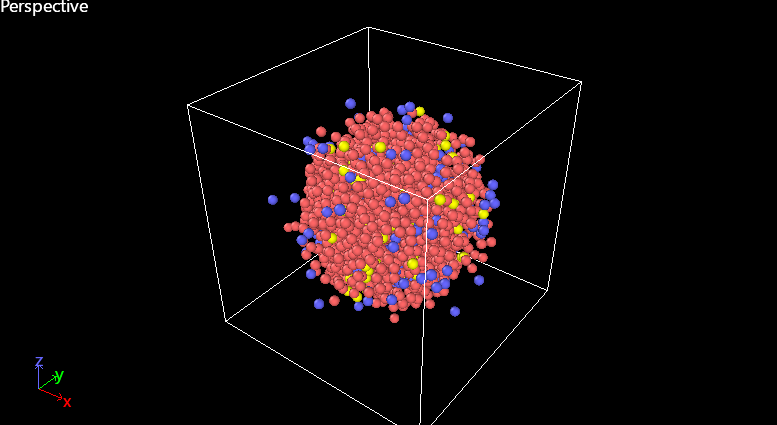
After running, the structure looks like this (screenshot attached).
My questions:
Does anyone know reliable LJ (or alternative) potential parameters for AlSi10Mg?
Am I correct to assume that the spreading issue is due to incorrect LJ parameters?
Would another potential type (e.g., EAM + LJ hybrid) be more suitable for this alloy system?
Any guidance would be highly appreciated!
Thank you,
Tasmiah Zaman