Hi everyone,
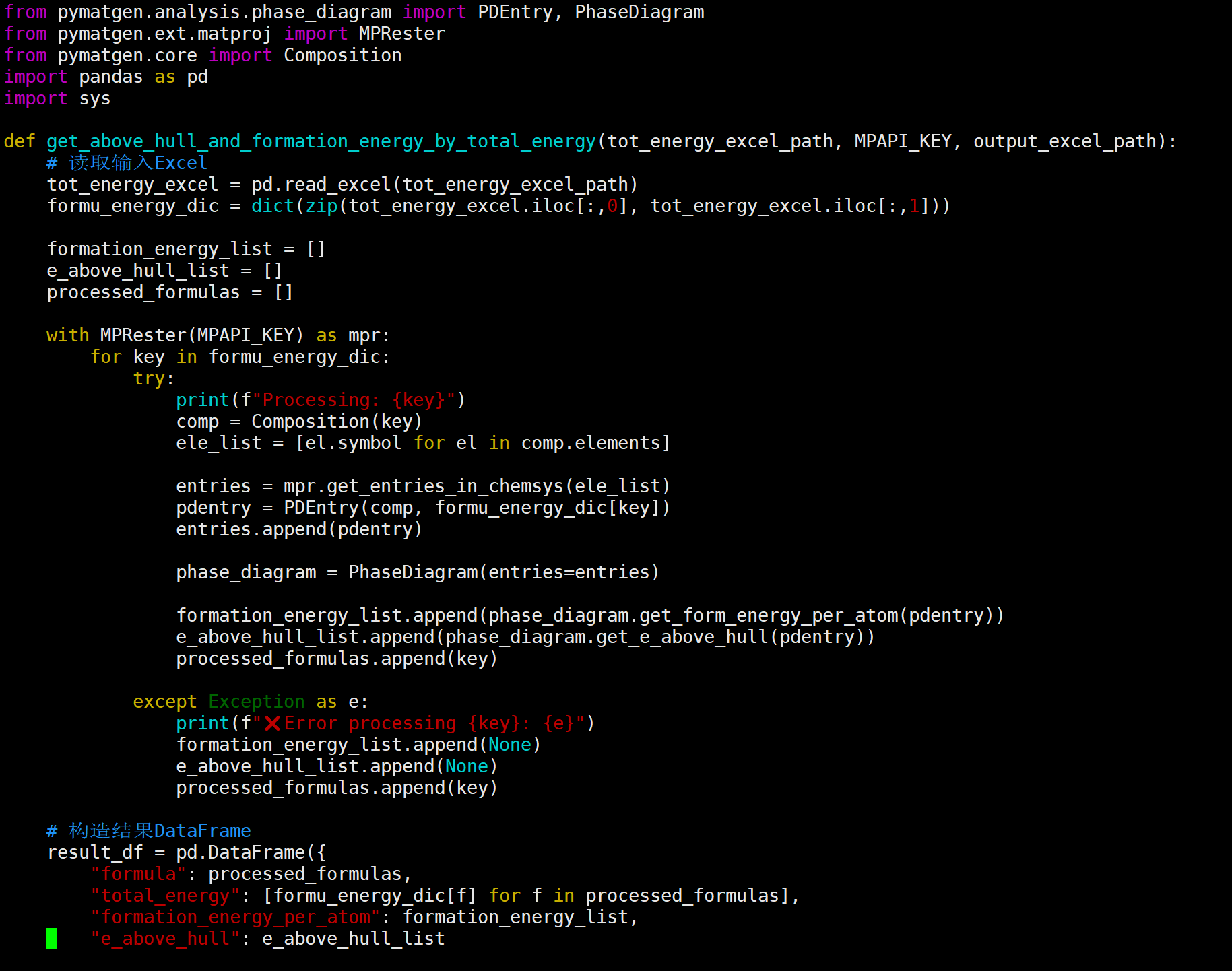
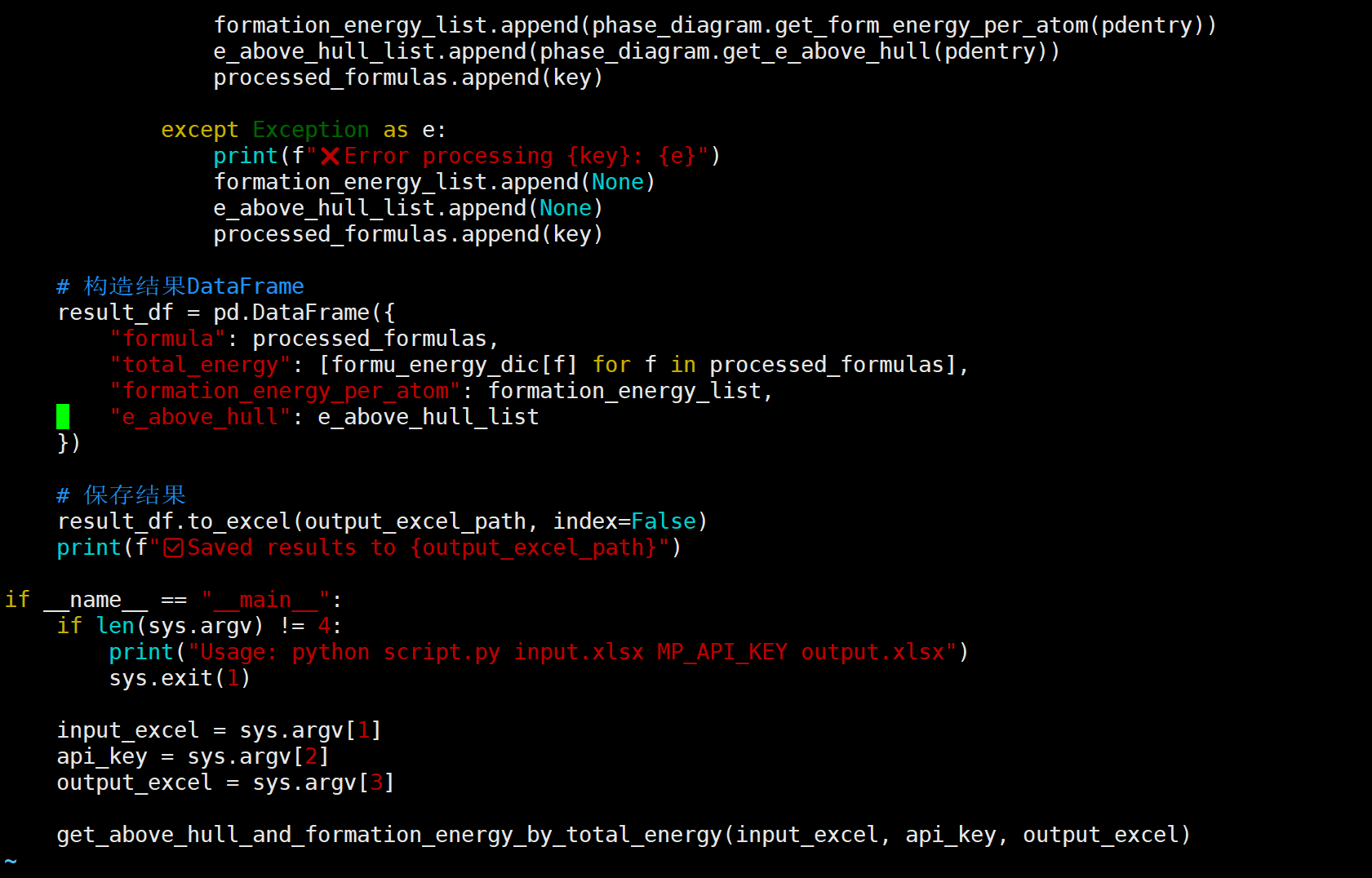
I’m currently working on calculating the energy above hull (Ehull) for a custom compound using the phase diagram module in pymatgen. I used DFT total energies from my own structure relaxations (VASP), and constructed the phase diagram based on the relevant chemical system.
However, I noticed that the Ehull values for my compound are consistently around 0.3–0.5 eV/atom, which seems unusually high.
I also tried using the total free energy values reported in the literature as input for the phase diagram construction in pymatgen, but the calculated Ehull values still don’t match those reported in the paper.
Any suggestions on how to reduce this discrepancy or debug this issue would be much appreciated!
this is my code,
Thanks in advance.
Are you sure that the DFT functional you’re using accurately describes this system? Determining the right functional, and possibly Hubbard U corrections is important for electronic structure calculations
Thank you very much for your response and helpful comment.
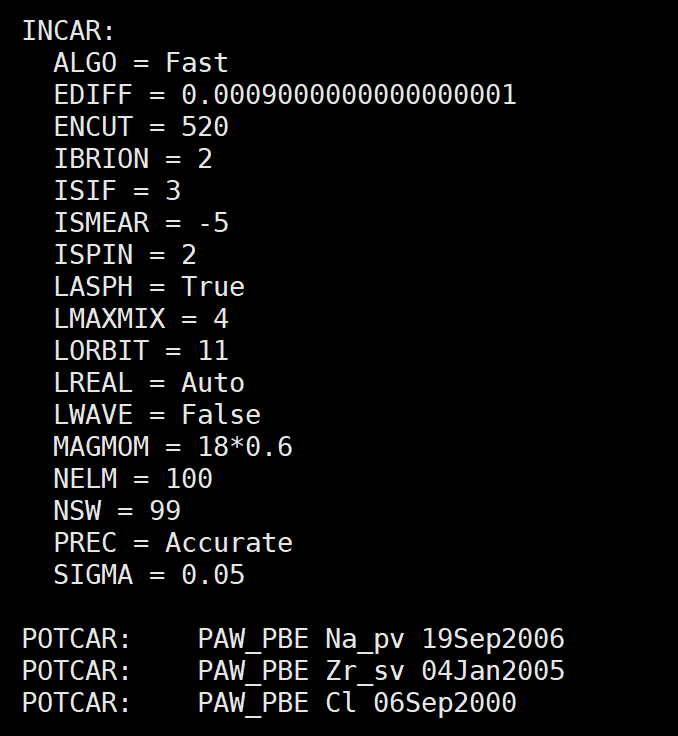
At first, I set the VASP input parameters manually. Later, after reading several forum posts related to Ehull calculations, I switched to using the MPRelaxSet module from pymatgen to generate the input files.
The structure I’m working on is Na₂ZrCl₆, which is not available on the Materials Project. According to literature, it adopts a P2₁/c space group. Since no direct structural file was available, I built the POSCAR by modifying structures from Na₃ErCl₆ and Na₂ZrF₆ as templates.
Based on the OUTCAR, I confirmed that the calculation uses the GGA-PBE functional. I did not apply any Hubbard U corrections, as the system does not contain strongly correlated electrons.
In addition, I found a publication reporting the Ehull of Na₂ZrCl₆ is about 25 meV/atom, which supports its metastable nature.
Please let me know if you have further suggestions or if there’s anything I might have overlooked. I appreciate your feedback.
Best regards,
fei
If you use spglib or pymatgen to get the space group of the material, e.g.:
from pymatgen.core import Structure
for file_name in ("POSCAR","CONTCAR",):
print(file_name, Structure.from_file(file_name).get_space_group_info())
do you see that the expected P21 / c symmetry is respected before and after relaxation?
To build the phase diagram, are you using entries you computed yourself for the unaries and other composite phases? Or the entries from Materials Project / mp_api?