Hello everybody ,
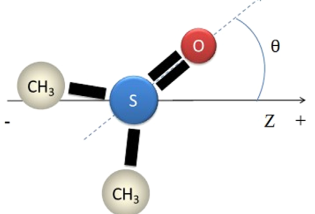
There is a DMSO molecule that is shown below . I want to compute the orientation of DMSO by using lammps , which is defined by the angle θ between the vector SO (from the sulfur atom pointing to the oxygen atom) and the z-direction . Can I compute the angle θ with lammps ? How do I set it up ?
I would appreciate it if anyone could give me some advice .
with regards!