Dear LAMMPS users and developers,
I’m currently trying to describe the Tersoff potential and the Coulomb interaction together.
I know that I can use both pairs styles together through hybrid/overlay, but what I want to know is to specify the minimum distance that the Coulomb interaction applies.
For example, I want to apply the Coulomb interaction only when the distance between atoms is greater than 2.5 angstroms while applying the Tersoff potential for all atoms.
I kept looking for the LAMMPS command, but I couldn’t find the minimum distance to exclude that potential.
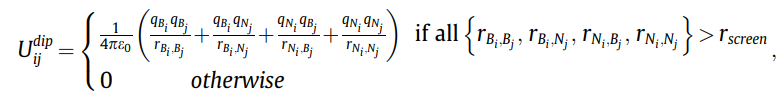
I would be really grateful if I could get your help.
Thanks.