The methodology docs on dielectric calcs state that MP uses a k-point density of 3,000 per reciprocal atom and a plane wave energy cut-off of 600 eV.
The 2nd Atomate dielectric paper further recommends setting EDIFF to 1e-6:
…we believe that parameter set 7 is a good balance between efficiency and computational convergence and hence was chosen for comparison of DFPT results with experimental data.
Those were the settings we’ve been running all our DFPT calculations so far (just a few hundred for now). We initially checked only that we reproduce the MP refractive index with those settings.
Last week we realized to our surprise that we get quite different results for the ionic contribution for the dielectric tensor.

This prompted us to check if our calculation settings were in fact the same as what MP uses. Turns out that has no DFPT calculations using the settings listed in the methodology.
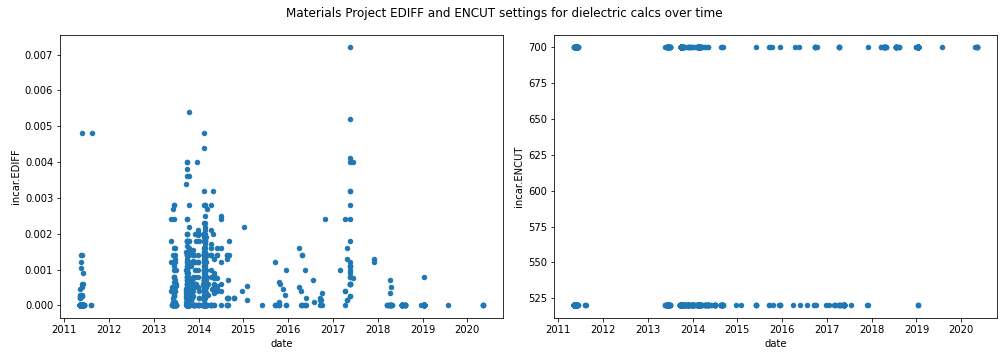
Instead, ENCUT is either 520 or 700 eV and EDIFF all over the place.
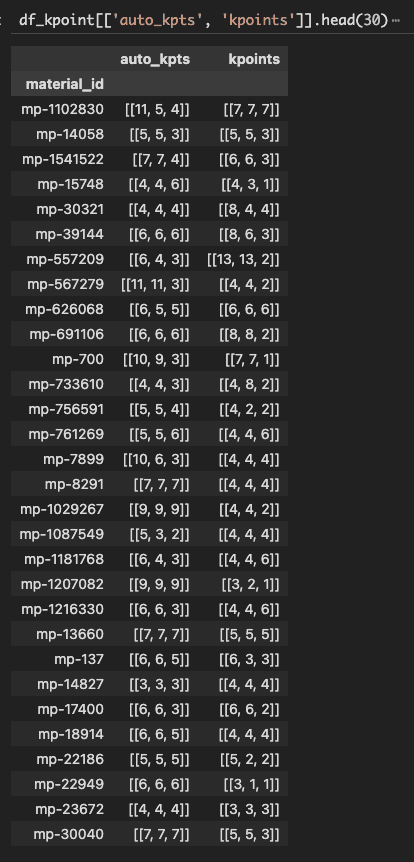
Similarly, there’s discrepancy between the k-points output by Kpoints.automatic_density(struct, 3000) and what MP uses (the kpoints column in the below screenshot):
As we’d like to reproduce MP results as faithfully as possible, could someone let us know how you guys pick the settings for your DFPT calcs?