Hi, guys
I am running the EMD simulations of 5-site CH4 confined in (10, 10) CNTs,
using the Lammps version (10 August, 2015). I have a very particular and
fundamental question regarding how Lammps deals with the pair interactions
between two rigid molecules, i.e. when the cut off radius applies, is it
applied on the distance between the center of masses of two molecules or
any two individual atoms directly?
the force computation in LAMMPS knows nothing about how particles are time
integrated. thus it pays no attention to groups, molecules or rigid
objects. interactions are computed between particles on the particle
positions.
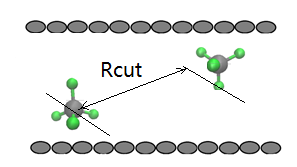
For instance, depicted in the Figure below, when the distance between the
center of masses of two methane molecules are within the cut off radius,
should we
(1) include the interactions ( L-J and Coulomb) for all the pairs of
atoms belonging to these two molecules, although most of the individual
pair of atoms are out of the cut off radius;
(2) or only considered the pairs of atoms are located within the
cutoff radius?
!image001.png|297x167
In the first case we guarantee the electrical neutrality of the methane
molecules, while the second case breaks this fundamental rule and generally
induce unexpected high interactions (inter molecular forces) between these
two molecules. I learnt from the Lammps manual that, Lammps follows the
second case to utilize the cut off to handle the L-J and Coulomb
interactions for rigid molecules. However, I cannot convince myself this is
a correct way to do this, and since this is so fundamental I hope you guys
can help me out of this.
what LAMMPS does is consistent with how pretty much most MD codes work
these days. what you describe is similar to (but not exactly the same), as
what is called "charge groups" in some MD codes, where the following rule
applies: if one atom of a charge group is inside the cutoff, interactions
for all atoms are computed. this is an important consideration, when one
uses coulomb interactions with a cutoff to mitigate the impact of the
rather drastic truncation happening. however, this has become irrelevant
due to using kspace solvers to compute the *complete* set of coulomb
interactions for the entire system (and infinite periodic replication)
through ewald summation and similar methods. through the used of the
damping function in real space, there is no significant charge interaction
left after the (per atom pair) cutoff is reached.
for LJ interactions, this is all much less of a concern, as the LJ
interaction decays much more rapidly, however, LAMMPS offers pair styles
and kspace solvers to treat the attractive LJ term via an ewald summation
approach. for the kind of system you describe, a suitable real-space cutoff
should suffice. it is easy to test for convergence. i would expect, that
something in the range of 12-15\AA will do.
axel.
{kind=link}