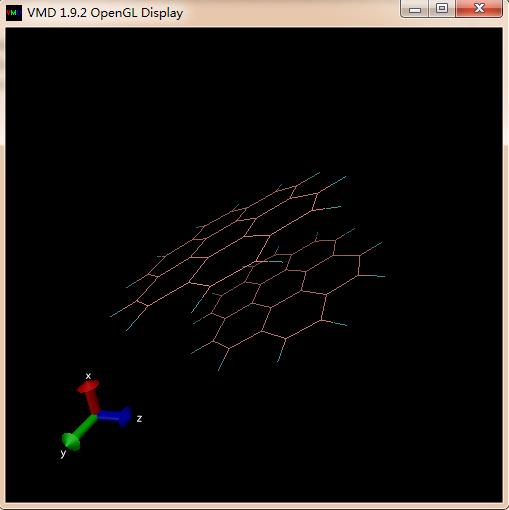
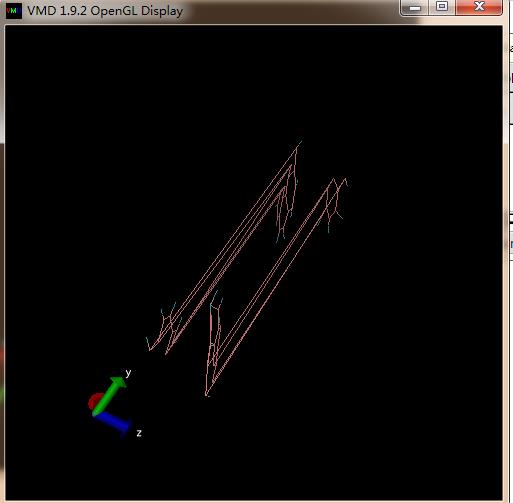
Dear lammps users
I have used Reaxff forcefiled in LAMMPS to perform MD simulation and I want to observe bond breaking and formation during the simulation. I heated the system to 1800 K with the fixed heating rate to see whether there is bond breaking. I got the trajectory from the lammps, and then I exported the trajectory to VMD to observe the bond breaking. However, I got strange results. The first picture is the initial structure; the second picture is the structure from specific temperature. It seems that there is bond breaking in second one, but it looks so strange. My question is
(1) Whether VMD can be used to analyze results from Reaxff forcefiled in LAMMPS
(2)If not, is there other tools can be used to analyze results from Reaxff forcefiled in LAMMPS
Sincerely
Fan Li