Hi everyone, I am using reaction force field during lammps simulation and want to apply electric field to the system. But I can only apply an electric field of a fixed size in a fixed area by the command Apply command is as follows:
region upp block INF INF INF INF INF 23.5555 26.5555 units box
group upp region upp
fix 1 upp efield 0.0 0.0 0.3 region upp
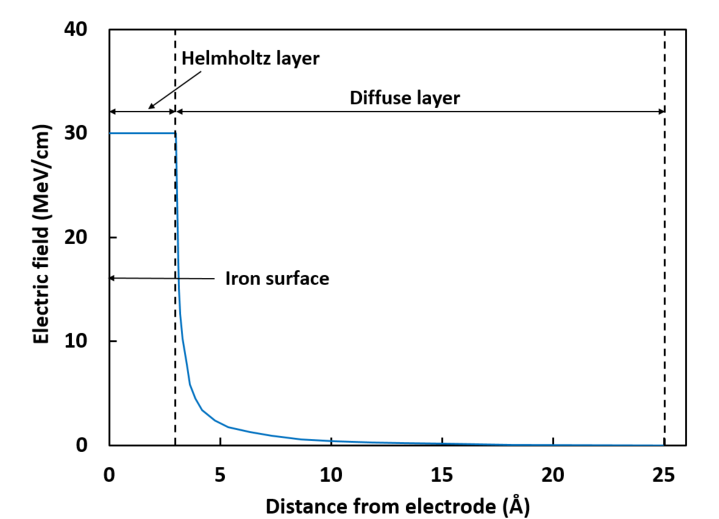
However, as I can see from reading the literature, the authors of the literature apply what appears to be a variable electric field, as follows:
Having said that, there’s good theoretical reasons why it’s possibly not correct to apply such a crude continuum-model electric field in an electric double layer simulation. There’s a much simpler and more exact method to be used with the same recent LAMMPS code.
I’d be interested in working together on a paper to test if the simpler method works – email me at [email protected] if you’re interested.
Thank you very much for your excellent contribution to the lammps community!
I would like to ask what kind of command to use based on the current lammps software to achieve such an electric field application? According to the literature, this is the form of electric field applied by the authors, with some results. Is it necessary to modify the source code to apply the electric field in this way?
As the above documentations state, this feature is present in the 15Jun23 version of the code. Please search for the “LAMMPS build instructions” if this version is not compiled on your computer or cluster (which it almost certainly isn’t) and talk to your system administrator about how to build it properly.
The literature contains all kinds of results with all kinds of quality, from benchmark-worthy studies to complete wastes of electricity. I am quite sure the method you described is wrong (the method’s fault, not yours), but you shouldn’t take my word for it – I am just a stranger on the Internet and my opinion does not determine whether simulations match up to reality (or a reviewer’s judgement).
Please note that if you are applying (1) a ReaxFF simulation (2) to a heterogeneous phase, electrode-electrolyte system (3) with applied potential, your simulation has three separate sources of complexity, and if you are not experienced with molecular dynamics you run the risk of getting plausible-looking but rubbish results. Please make sure you have a local mentor who is at least experienced in molecular dynamics, if not one of these specialised techniques.