Hi,
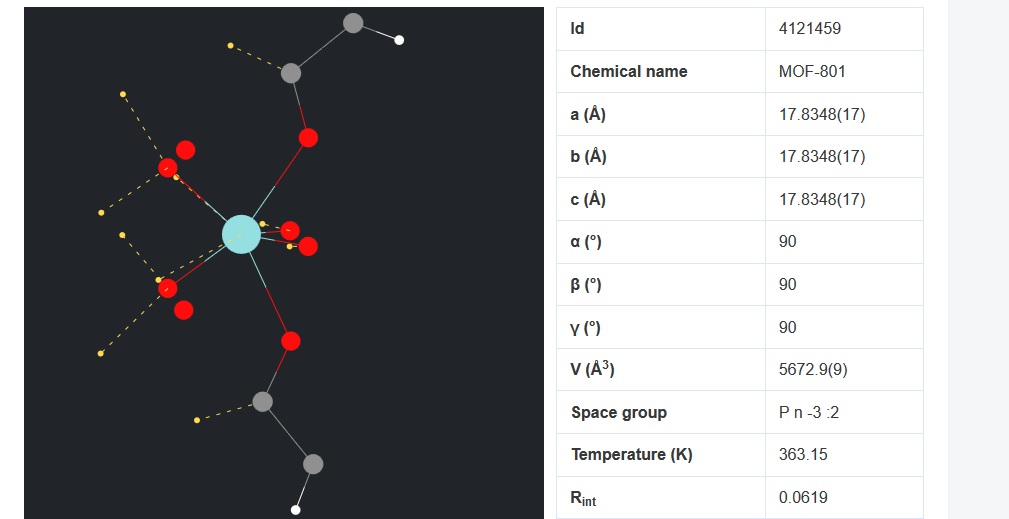
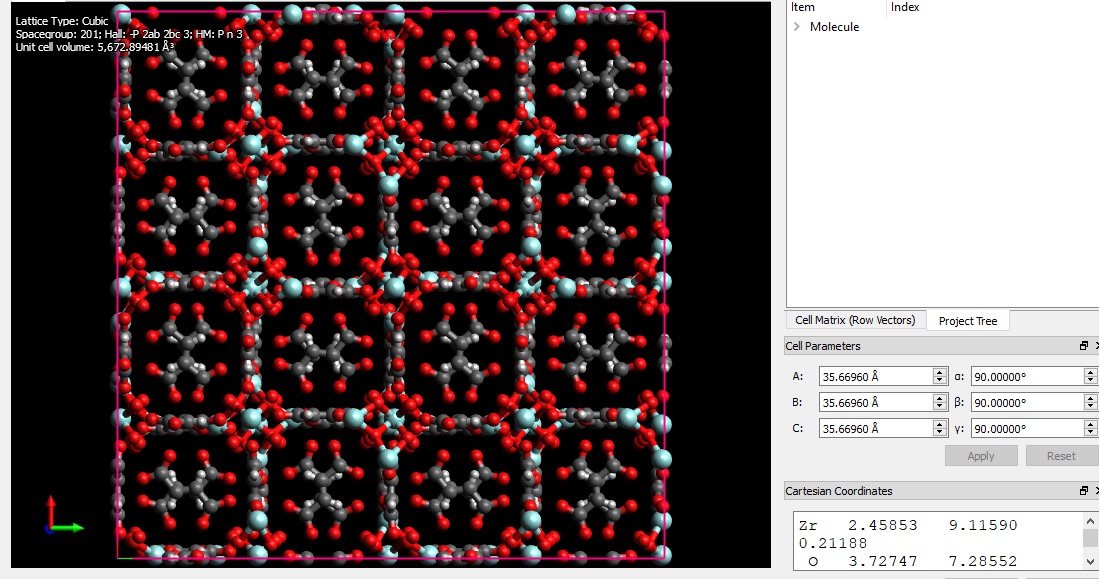
I want to simulate a MOF structure using LAMMPS and need a structure file of the MOF. I found some cif file (fig 2) of the MOF unit cell and open them using visualization softwares such as VMD and Avogadro (figure 1). But, there is a problem about cross boundary atoms. some of the atoms are in the negative values and there will appear from the other side of the box. By replicating the structure and building the supercell (figure 3), that part of the structure seems to be in complete and there are some free atoms without any bonds. how can I handle this problem? I searched this problem and found some answers which say this is not a concern in LAMMPS simulation. Is it true?
Thanks
Probably what they meant is that this is not a LAMMPS question. If you have the wrong connectivity, you will have problems in your simulation, be it carried out with LAMMPS or any other software.
On what concerns your question: if I understood correctly, the thing is that the atoms sitting at the opposite edges of your cell should be bonded. You can easily declare this bond by yourself by building a python code. Here goes an example of a python code part which can be used to tackle the issue. In the example, A is an array containing 5 columns: atom_ID, atom_type, x, y and z of all the atoms in your system. I am assuming the array is ordered in such a way that the blue atoms in your figure (Zr, I suppose) appear first and the red (O, I suppose), appear just after. The variables i, j, k are the vectors generating your cell, which is assumed to be cubic (given the figures you showed) and mod_i, mod_j, mod_k are the magnitude of the vectors, respectively. The “if” conditions will ensure that you always consider the given pair of atoms in the closest distance possible, given that the system will be treated with periodic boundary conditions during the simulation and thus you will have an image of them. So if a given atom in your simulation box is bonded to the image of another one, you will be able to realize this with the setup below. If you do some thinking on these “if” conditions, you will see how/why it is working in the direction I previously mentioned. Finally, the last if condition in the code is ensuring that you declare the given pair of atoms as bonded if indeed they (their images included) sit at a distance closer than the bond length. The threshold should be small and simply account for (a) floating errors and (b) the possibility that your structure is not ultra perfect to the point where everything is sitting at the rightful relative distances.
for it_1 in range (0, N_blue):
for it_2 in range (N_blue, N_blue + N_red):
dist_x = A[it_1,2]-A[it_2,2]
dist_y = A[it_1,3]-A[it_2,3]
dist_z = A[it_1,4]-A[it_2,4]
if (abs(dist_x) > abs(((mod_i)/2))) & (dist_x > 0) :
dist_x = dist_x - i[0,0]
if (abs(dist_x) > abs(((mod_i)/2))) & (dist_x < 0):
dist_x = dist_x + i[0,0]
if (abs(dist_y) > abs(((mod_j)/2))) & (dist_y > 0) :
dist_y = dist_y - j[0,1]
if (abs(dist_y) > abs(((mod_j)/2))) & (dist_y < 0):
dist_y = dist_y + j[0,1]
if (abs(dist_z) > abs(((mod_k)/2))) & (dist_z > 0) :
dist_z = dist_z - k[0,2]
if (abs(dist_z) > abs(((mod_k)/2))) & (dist_z < 0) :
dist_z = dist_z + k[0,2]
if dist < bond_length + threshold:
temporary_array = np.array([[bond_ID, bond_type, A[it_1,0], A[it_2,0]]])
bonded = np.concatenate((bonded, temporary_array), axis = 0)
Dear Cecilia,
Thanks a lot for your reply. I will try it and it should work.
Cincerely
Dear Cecilia,
Thanks again for your reply. I’m a beginner in LAMMPS and molecular simulation and sorry for elementary questions. If I am right, the python script you’ve sent should be apply to the MOF data file before the simulation. Should I apply the changes to the data file and then replicate the unit cell in the LAMMPS script? I found this unit cell data file from the net and made a supercell (222) by Avogadro (the 3rd fig in the first post). I found some other data files of this MOF from the supporting information links of related papers. Both structures (made supercell and data file from published papers) seems to be incomplete. I suppose the reason is due to the edge atoms which crosses the unit cell wall.
Yes
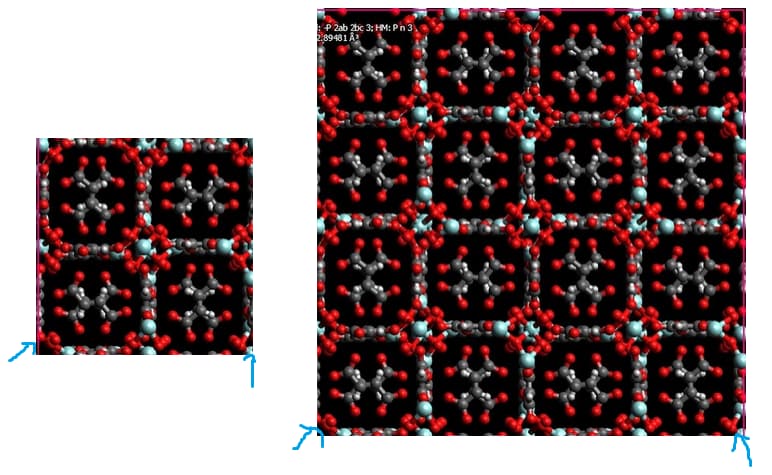
So, you first create the simulation box you are going to actually use in the simulation, which should correspond to the desired amount of unit cells propagated in each direction (in your case a 2x2x2 I suppose). Then you run the code to create the connectivity. You should not create the connectivity before propagating the unit cell otherwise you will be declaring bonds and angles that do not actually exist. Imagine for example if you were creating the bonds using the simulation box shown in the figure in the left. You would be declaring the atoms I indicated in the blue arrow as bonded… when actually the bonded ones are the ones from the figure in the right.
PS: I suppose you got that when I said in my first answer “i, j, k are the vectors generating the cell” I meant the simulation cell (or simulation box) and not the unit cell. So this changes accordingly to the size of the simulation box you are building from propagating the unit cell
Thanks a lot for the reply. I got the point you noted, but still one problem remains. as the figure I uploaded here, the green bonds should exist in the final structure which are missed in our structure. It seems that this is because of the cross boundary atoms which considered as non-bond. the red atoms should bind to blue one. I think if I replicate the structure and then use the python code, these bonds will be missed.
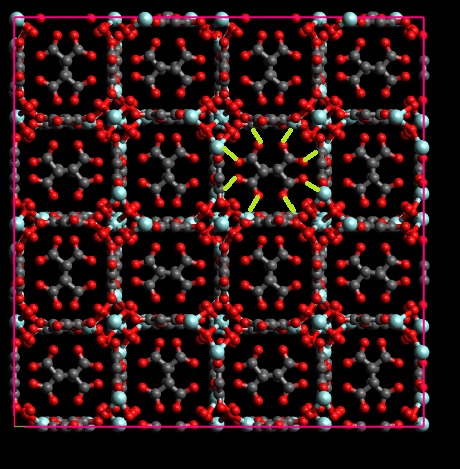
Ahh, but these are not really bonds across the periodic boundaries in your simulation box (I thought your problem was related to that).
Well, you can still solve this with python coding in different ways. If your xtal structure is close to “perfect” (i.e., every atom sitting at a close-to-ideal distance from one another), you can easily find the bonds between the metal ions and the oxygens by using the piece of the code I gave you: all the oxygens that should be bonded to the metal will meet the criterium dist < bond_length + threshold previously mentioned.
If somehow you would have other oxygens that should not be bonded to a given metal also meeting this criterium, you could make a more sophisticated code that is able to differentiate the rightful bonded oxygens by some other criterium.
PS: there is also this create_bonds command in LAMMPS that you can try using if you are having problems with coding (create_bonds command — LAMMPS documentation). I am not familiar with it (never used). From reading the first sentence it seems that the underlying idea of the command is also based on distance between atoms.