Dear LAMMPS users,
Hi,
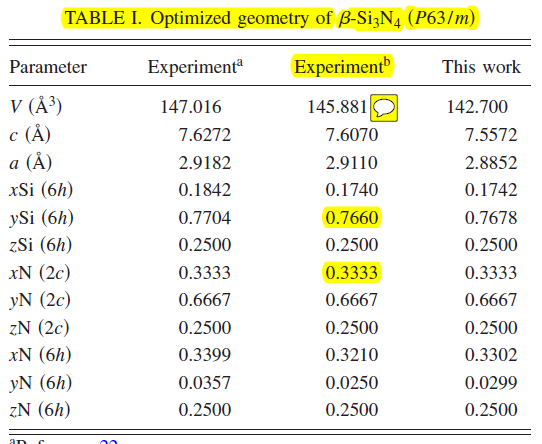
Currently I intend to build an initial lattice structure of Silicon nitride. The atomistic details based on an experiment is attached and I used the input commands as below. however, when I run this job it doesn’t result a correct structure ( It gives something but It isn’t correct ). So, I wonder if you could please help me out. By the way, I studied “http://lammps.sandia.gov/doc/Section_howto.html#howto-12” so please don’t reference me into that webpage.
Sincerely,
Ali
6. How-to discussions — LAMMPS documentation
lammps.sandia.gov
6.1. Restarting a simulation. There are 3 ways to continue a long LAMMPS simulation. Multiple run commands can be used in the same input script.
|
- |
---------- Initialize Simulation ---------------------
units metal
atom_style atomic
dimension 3
boundary p p p
#------------Define variable------------------------
variable a equal 7.5572
variable b equal 7.5572
variable c equal 2.8852
variable gamma equal “120.003/180*PI”
variable singamma equal “sin(v_gamma)”
variable cosgamma equal “cos(v_gamma)”
variable bx equal “v_bv_cosgamma"
variable by equal "v_bv_singamma”
#------------Define Si3N4 lattice------------------------
lattice custom 1.0 &
a1 a 0.0 0.0 &
a2 {bx} ${by} 0.0 &
a3 0 0.0 $c &
basis 0.3333 0.6667 0.2500 &
basis 0.3302 0.0299 0.2500 &
basis 0.1742 0.7678 0.2500 &
orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
#------------------Create Si3N4 nanowire--------------
region zona1 prism 0 1 0 1 0 1 -0.37 0 0
box tilt large
create_box 2 zona1
create_atoms 2 box &
basis 1 1 &
basis 2 1 &
basis 3 2 &
Atom 1 = N and Atom 2 = Si
mass 1 14.01
mass 2 28.085
write_data Ali.in