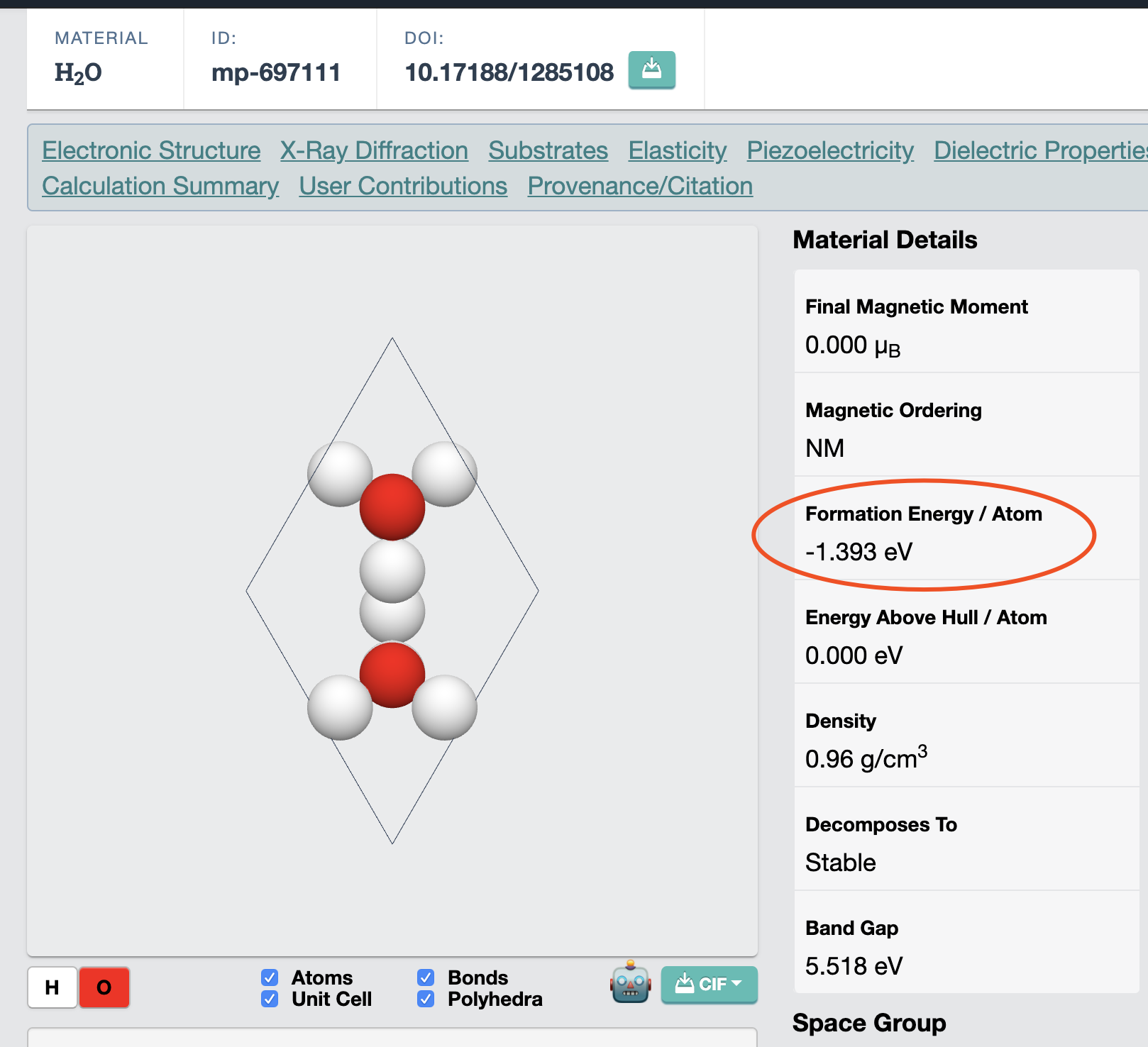
In mp database, I found the formation energy for water molecules(mp-697111) should be -1.393 eV*3=-4.179/formula.
However, if I search the Final Energy/Atom for H2O(mp-697111), H2(mp-632291) and O2(mp-1091399), each of them is with the smallest e above hull and green highlight. Then I calculated the formation energy for H2O by hands:
Where does the error come from? Similarly, I calculated the formation energy for Mg(OH)2, I found there is also a deviation between the data in MP and data calculated by hands.
This troubles me for a long time if you have any idea, please leave a message, Thanks in advance.
Thanks for your reply!
I know formation energies are in eV/Atom and I calculated the formation energy for the water molecule, -1.393*3 = -4.179 eV/formula.
Mg question is If I search the energy of H2,O2 and H2O in mp database, then I calculate the formation energy by hands(the answer is -3.1765/formula), where this deviation(between -4.179 and -3.1765) come from?
Hello, when i use the corrected energy and your equation, I calculated the formation energy by hands , then it have the result is -135.2761 eV. Can you tell me what my problem? Thank you so much
I do not understand the formation energy calculation. EH2O (corrected energy) is -62.3130 eV. (-62.313+6.78+0.5*9.857) = -50.6045 eV. Dividing it by 3 does not produce the result. Please help me to understand.
Sorry @Tanmoy_Paul for the late reply. The issue of calculating formation energies correctly comes up very often, so I wanted to clear it up for anyone who stumbles upon this in the future:
When calculating formation energy by hand, you have to be extremely careful about energy normalization. When you have the energy result of a DFT calculation, there are three possible energy normalization schemes you could follow:
Normalize the energy to a single atom (this is the standard eV/atom you see. In this framework, H2O effectively becomes H_{0.333}O_{0.666})
Normalize the energy to a single formula unit (i.e. say your calculation of H2O contains 12 atoms → this would equal 4 formula units of H2O)
Normalize the energy to the total number of atoms in the cell of the calculation (i.e., don’t divide the DFT output by anything)
When you write the equation for formation energy, you can choose to either do it with chemical potentials, which are already normalized by scheme #1, or with a reaction equation as has been done in this post (scheme #2). Just make sure that whatever method you pick has consistent normalization throughout. For the H2O example discussed here, this would be:
ΔE_f(H_2O) = E(H_2O) - E(H_2) - 0.5*E(O_2)
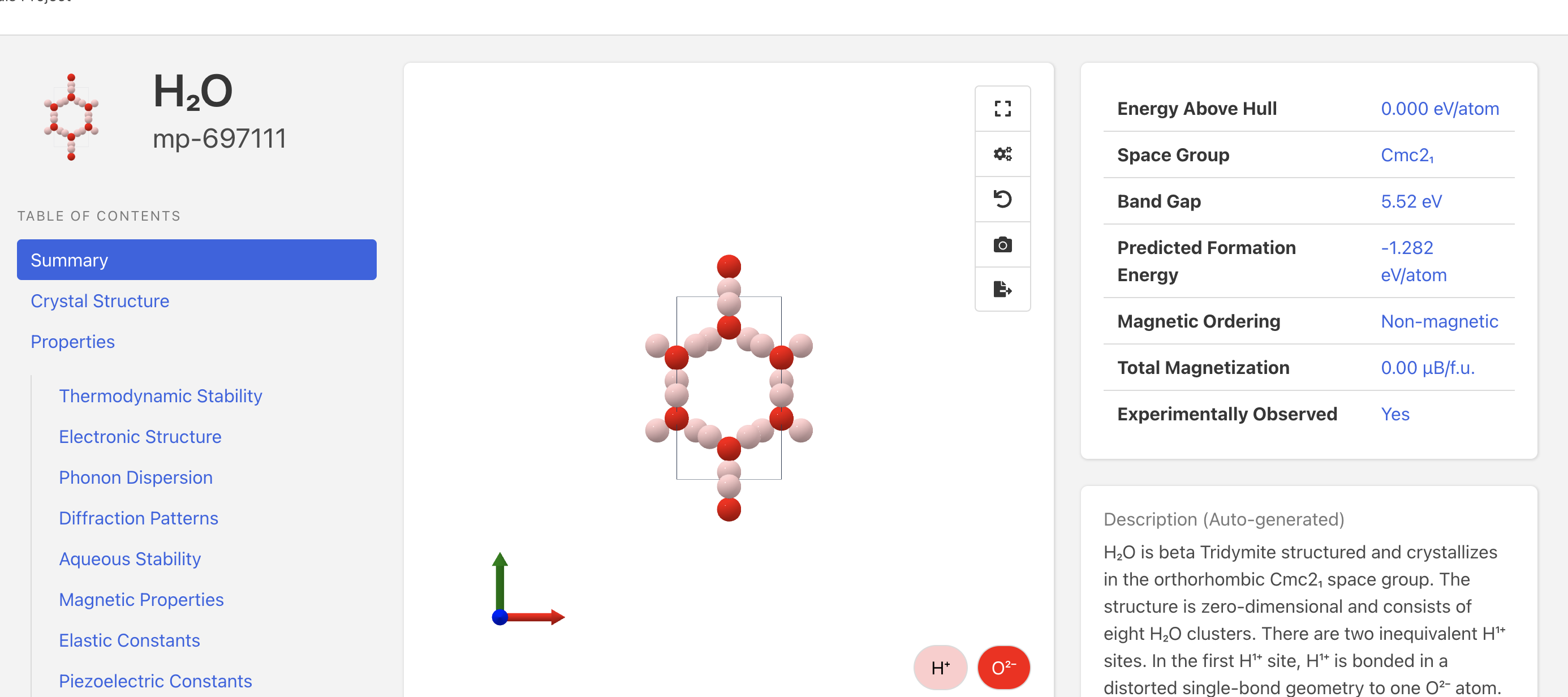
If you are using the Materials Project, you will get total energies for the entire calculation cells. The data below is from the new MP website and API:
E(H2O) (12 atom cell) = -62.3132 eV → (divide by 4 to get -15.578 eV/f.u.)
E(H2) (4 atom cell) = -13.5709 → (divide by 2 to get -6.78545 eV/f.u.)
E(O2) (8 atom cell) = -39.5735 eV → (divide by 4 to get -9.893 eV/f.u.)