Hello EMC,
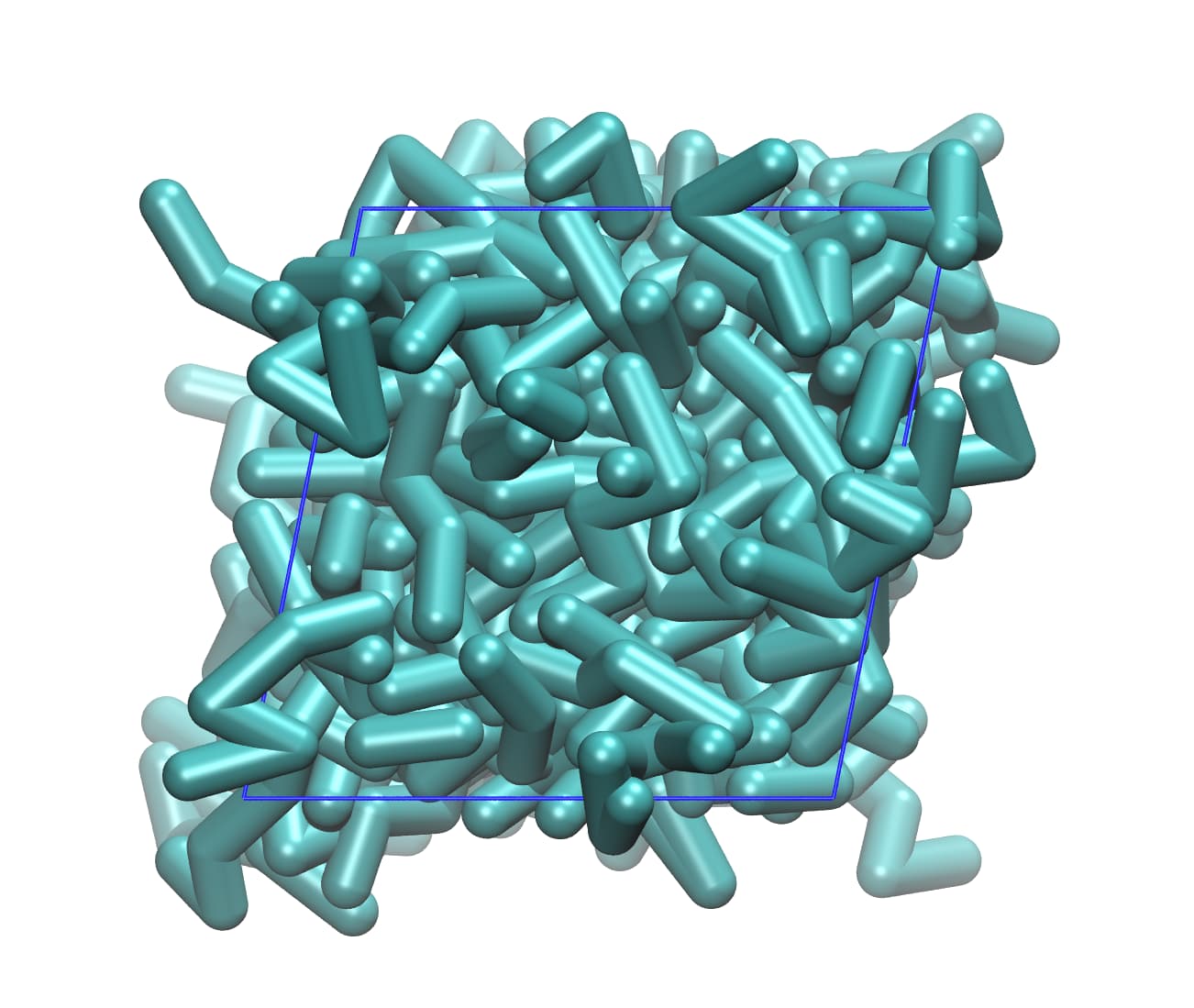
Currently, I am building the semi-crystalline PE with chain stems titled so that the {201} crystallographic plane is normal to the z direction. I made the system (4 * 6 * 100) like below
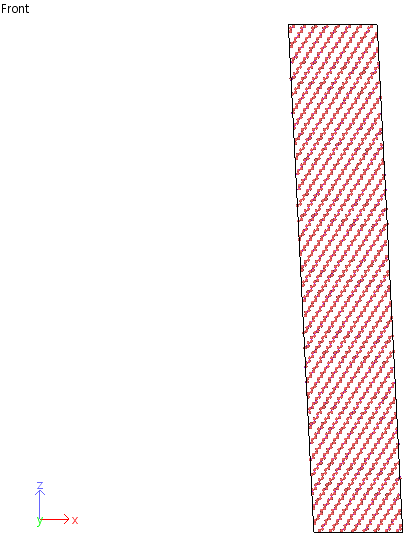
However, I want to change the angle of simulation box, beta, to be 89.8 degree.
I guess the “deform command” in 5.7 (docs) should be used.
Is there any advice you can offer?
Below is my build.emc for semi-crystalline PE.
(* create a semicrystalline polyethylene input structure by using a
crystalline polyethylene unit cell *)
(* set variables *)
variables = {
input -> $root+"lib/polyethylene.emc",
output -> "pe",
ncycles -> 100,
nblocks -> 1,
seed -> 1
};
(* load unit cell *)
get = {name -> input};
(* set random seed *)
simulation = {units -> {seed -> seed}};
(* create a supercell, using a 201 crystal facet cut, based on a 001
coordination *)
crystal = {n -> {4, 6, 100},merge -> true,periodic ->{false, false, false}};
(* immobilize the crystalline region 100% crystallinity*)
flag = {flag -> fixed, oper -> set,
region -> {shape -> cuboid, center -> {0, 0, 1/2},
radius -> {1, 1, 50/100}}};
(* cut 0 polymer chains within a selected region to accomodate PE molecular
mass *)
cut = {n -> 0,
region -> {shape -> cuboid, center -> {0, 0, 0},
radius -> {1, 1, (1-6/9/5)/2-1/5}}};
(* remove a maximum of 0 end beads within a selected region to accomodate
the target amorphous density and PE molecular mass *)
remove = {n -> 0, mode -> cluster,
region -> {shape -> cuboid, center -> {0, 0, 0},
radius -> {1, 1, (1-6/9/5)/2-1/5}}};
(* terminate all chains with the apropriate termination methyl group *)
terminate = {mode -> all};
(* set system *)
simulation = {
types -> {
applied -> true
},
systems -> {
properties -> {
{id -> 0, t -> 10000}}},
moves -> {
displace -> {frequency -> 51}, (* specify move frequencies *)
rebridge -> {frequency -> 14},
reptate -> {frequency -> 7},
endbridge -> {frequency -> 14},
rotate -> {frequency -> 14}},
types -> {
virial -> false, (* do not calculate virial *)
neighbor -> sector, (* use sectors to find neighbors *)
stencil -> standard, (* use standard stencil *)
skin -> 0.0, (* use no skin *)
standard -> {
correct -> {active -> false}}}}; (* add long range correction *)
(* check force calculations *)
force = {style -> init, message -> true};
force = {style -> list, message -> true};
(* check memory usage *)
memory = {style -> full};
(* execute a monte carlo simulation for ncycles *)
run = {ncycles -> ncycles, nblocks -> nblocks, cycle -> 0};