Dear all,
Could you please explain for me how to create initial data for creating an Al2O3 amorphous by lammps?
Dear all,
Could you please explain for me how to create initial data for creating an Al2O3 amorphous by lammps?
Please note that is not really a LAMMPS specific question but a question about general MD simulation procedures.
Amorphous structures for MD are typically created by melting a bulk crystal structure and then quenching and relaxing it.
Crystal structures can be created with the lattice and create_atoms commands or imported from a data file with read_data.
I know what steps for creating of Amorphous structures are:
#####################################
units metal
atom_style atomic
read_data Al2O3.data
replicate 2 2 2
velocity all create 5000 87287
pair_style eam/alloy
pair_coeff * * AlO.eam.alloy Al O
neighbor 2.0 bin
neigh_modify every 1 delay 0 check yes one 1000 page 2000
fix 1 all nvt temp 5000 5000 0.1
fix 2 all deform 1 x final 0 15.70866 y final 0 14.29263 z final 0 18.1387987 units box
thermo 50
thermo_style custom step cpu temp press ke pe etotal vol
thermo_modify lost ignore
dump 1 all atom 1000 dump.lammpstrj
run 500000
unfix 2
unfix 1
fix 1 all nvt temp 5000 3000 0.1
run 500000
unfix 1
fix 1 all nvt temp 3000 3000 0.1
fix 2 all deform 1 x final 0 14.280600 y final 0 12.993300 z final 0 16.489817 units box
run 500000
unfix 1
unfix 2
fix 1 all nvt temp 3000 3000 0.1
run 500000
unfix 1
fix 1 all nvt temp 3000 300 0.1
run 1000000
undump 1
unfix 1
fix 1 all nvt temp 300 300 0.1
dump 1 all atom 1000 dump2.lammpstrj
thermo_style custom step temp density cpu
run 1000000
write_data new_data.txt
But my question is how to create the initial Al2O3.data by Lammps.
I used some software to create it but I want to use just lammps.
Or If there is a way to convert cif file to lammps data?
When you already know that, then why do you ask for it?
Answers to questions can only be as good as the questions. If you don’t provide any information about what parts of the steps required you already know, you have to expect that people answering will assume that you don’t know any of it. So please don’t complain about being told what you already know when you don’t mention that.
See my previous comment. You could have asked this right away.
Nevertheless, I already answered that question. You can build a crystal structure by using the lattice and create_atoms commands. Please see their documentation. How to apply this to your specific case is an exercise for you.
Please also note that LAMMPS is not designed to be a “one stop shop” for all things MD. It is very deliberately designed to by a simulation engine, so its abilities to pre- and post-process data are limited (although not as limited than it was at the beginning a couple of decades ago). For everything else, you need to use pre- or post-processing software. A few links are provided at Pre/Post Processing Tools for use with LAMMPS but that list is not complete (we are dependent on people suggesting/providing links, and - given the nature of the internet and individual projects - links can change and software be discontinued at any time).
Thanks a lot for your explanation.
Marzieh@ Do you have any idea how to create Fe2O3 slab in the LAMMPS?
Hi
Unfortunately no.
Dear Alex,
I tried to generate an Al2O3 amorphous as you see as following. The RDF plot for O-O has a noise in 3.75 Angstrom in comparison to other reported results. May I ask you have a look at my input file
conv-1620-supercell333.dat (76.7 KB)
:
units metal
atom_style charge
read_data conv-1620-supercell333.dat
group Al type 1
group O type 2
set group Al charge 1.8
set group O charge -1.2
velocity all create 300 87287
#velocity all create 1900.0 87287
kspace_style ewald 1.0e-4
pair_style born/coul/long 11.172287
pair_coeff 1 1 0.0029 0.068 1.5704 14.0498 0. #Al-Al
pair_coeff 2 2 0.010150686 0.214 3.2678 5.08402081 0 #O-O
pair_coeff 1 2 0.0075 0.16892 2.6067 20.74482 0. #Al-O
neighbor 3.0 bin
neigh_modify every 1 check yes
###############################
#This block is npt at 300 K (500ps)
################################
fix 1 all npt temp 300 300 0.1 iso 0.0 0.0 1.0
dump 1 all atom 1000 dump1.lammpstrj
thermo 10
thermo_style custom step temp press density cpu
log log_1.txt
run 500000
write_data new_data1.txt
write_restart new_res1.dat
undump 1
unfix 1
##################################
#This block is nvt at 300 K (500ps)
##################################
fix 1 all nvt temp 300 300 0.1
thermo 10
thermo_style custom step cpu temp density press ke pe etotal vol
thermo_modify lost ignore
dump 1 all atom 1000 dump2.lammpstrj
log log_2.txt
run 500000
write_data new_data2.txt
write_restart new_res2.dat
undump 1
unfix 1
####################################
#This block is anealing 300 to 500 K (500ps)
###################################
fix 1 all npt temp 300 5000 0.1 iso 0.0 0.0 1.0
thermo 10
thermo_style custom step cpu temp density press ke pe etotal vol
thermo_modify lost ignore
dump 1 all atom 1000 dump3.lammpstrj
log log_3.txt
run 500000
write_data new_data3.txt
write_restart new_res3.dat
undump 1
unfix 1
##################################
#This block is npt at 5000 K (500ps)
##################################
fix 1 all npt temp 5000 5000 0.1 iso 0.0 0.0 1.0
dump 1 all atom 1000 dump4.lammpstrj
thermo_style custom step temp press density cpu
log log_4.txt
run 500000
write_data new_data4.txt
write_restart new_res4.dat
undump 1
unfix 1
################################################
#This block is nvt at 5000 K (500 ps)
#############################################################
fix 1 all nvt temp 5000 5000 0.1
thermo 10
thermo_style custom step cpu temp density press ke pe etotal vol
thermo_modify lost ignore
dump 1 all atom 1000 dump5.lammpstrj
log log_5.txt
run 500000
write_data new_data5.txt
write_restart new_res5.dat
undump 1
unfix 1
###############################################
#This block is cooling (100ps)
########## ####################################
fix 1 all npt temp 5000 300 0.1 iso 0.0 0.0 1.0
thermo 10
thermo_style custom step cpu temp density press ke pe etotal vol
thermo_modify lost ignore
dump 1 all atom 1000 dump6.lammpstrj
log log_6.txt
run 100000
write_data new_data6.txt
write_restart new_res6.dat
undump 1
unfix 1
###########################################
#This block is npt equilibrate the system at RT (100ps)
####################################
fix 1 all npt temp 300 300 0.1 iso 0.0 0.0 1.0
dump 1 all atom 1000 dump7.lammpstrj
thermo_style custom step temp press density cpu
log log_7.txt
run 500000
write_data new_data7.txt
write_restart new_res7.dat
undump 1
unfix 1
##########################################
#This block is nvt at RT
#############################
fix 1 all nvt temp 300 300 0.1
dump 1 all atom 1000 dump8.lammpstrj
thermo_style custom step temp press density cpu
log log_8.txt
run 500000
dump 2 all atom 10 dump-Final.lammpstrj
run 10000
write_data new_data-Final.txt
write_restart new_res_Final.dat
###################################
#This block is compue RDF
###################################
compute 1 all rdf 100 1 1
compute 2 all rdf 100 2 2
compute 3 all rdf 100 2 1
fix 3 all ave/time 1 1 10000 c_1[] file RDF-AlAl.txt mode vector
fix 4 all ave/time 1 1 10000 c_2[] file RDF-OO.txt mode vector
fix 5 all ave/time 1 1 10000 c_3[*] file RDF-AlO.txt mode vector
run 100000
I don’t have time to debug people’s inputs.
Noise is usually the result of lack of averaging.
Hi,
Thanks for your reply.
I will ask again, perhaps my explanation about the problem was not clear.
I generated Al2O3 amorphous by applying a “melt and
quench” technique that employed classical molecular dynamics (MD) performing lammps.
I used this potential:
pair_style born/coul/long
I tried to compute the partial RDF.
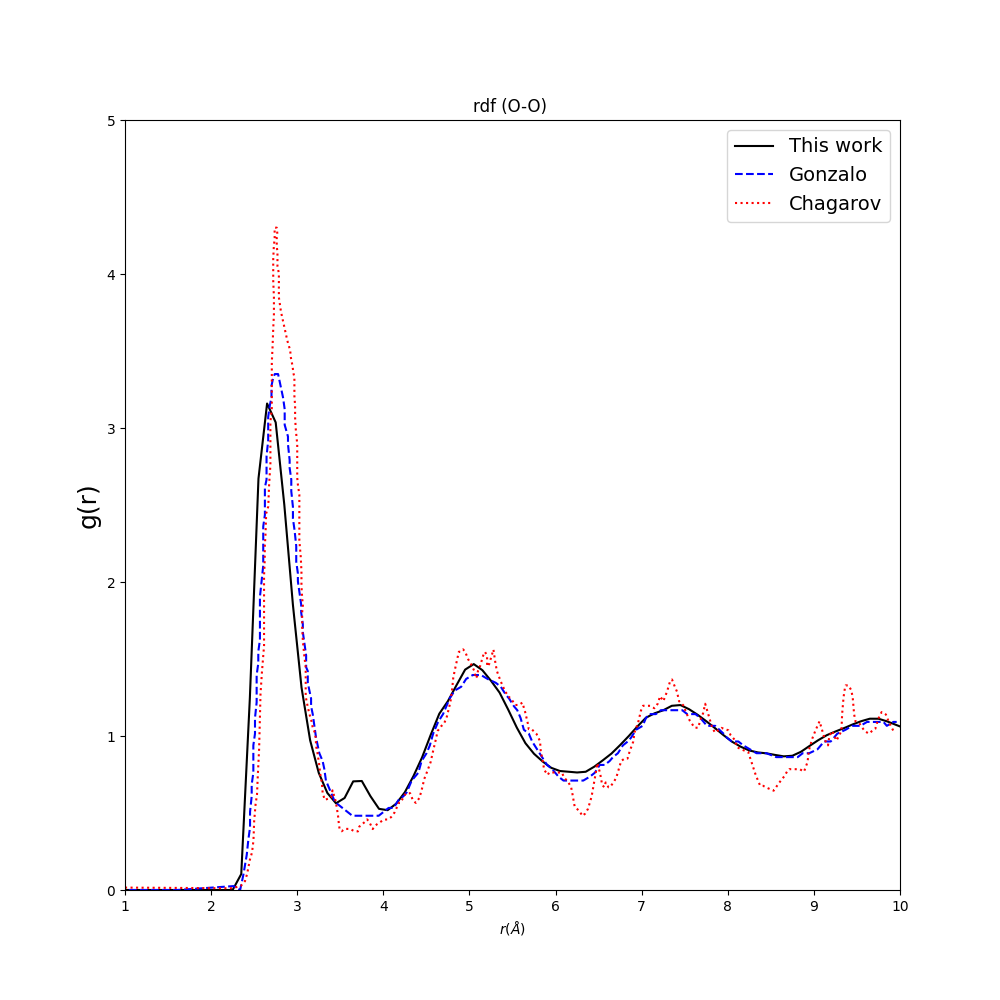
I have a small hump between 3.5-4 Ang in O-O (black curve is mine) in comparison to other reported results by Gonzalo(https://journals.aps.org/prb/abstract/10.1103/PhysRevB.65.104202) and Chagarov(https://iopscience.iop.org/article/10.1149/1.2986837/pdf) .
Why do I have this small hump? May I ask you how to solve it?
I mean what parameter or ensemble should I change/ modify to fix this problem?
Best,
Marzieh