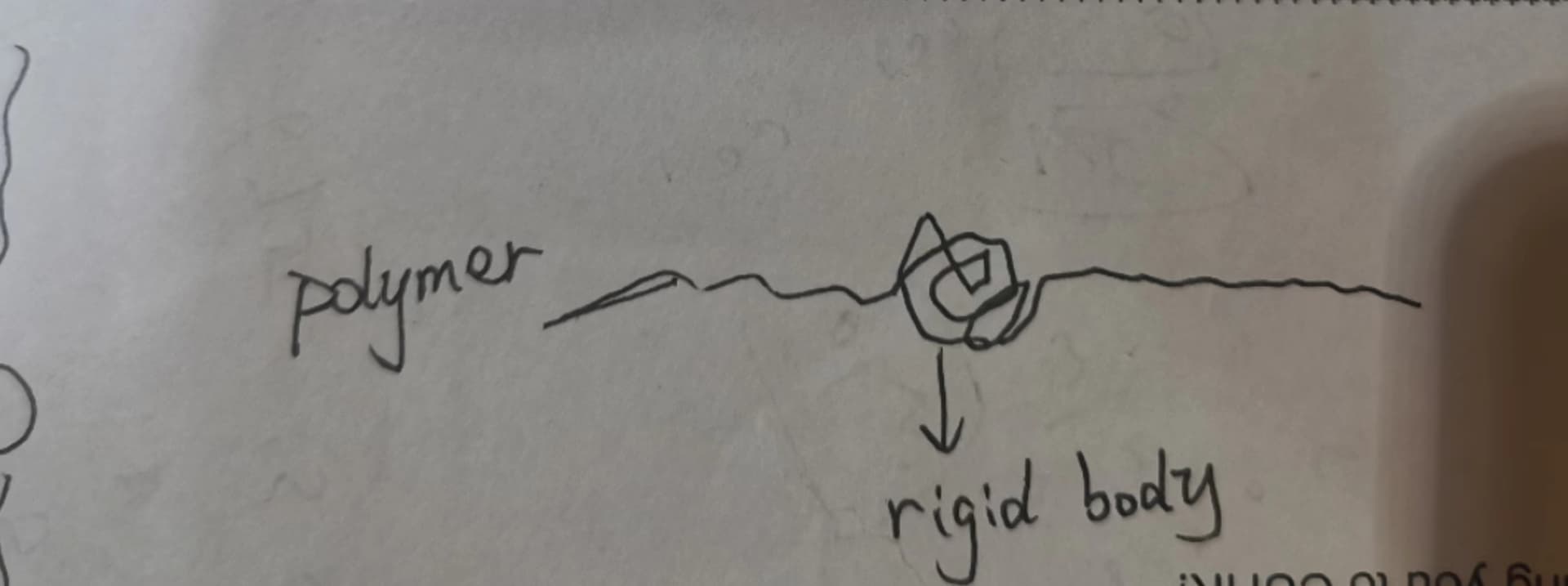
I need to make the center region of each polymer(40 in total) be a rigid body, which is approximately 30 beads long, and let the other part( on the two sides of the center region) flexible. Initially, I was trying to create groups and then “fix rigid” them, but it’s impossible to have 40 different groups in lammps and I cannot use “molecule” to definite rigid body since I want to leave part of each polymer flexible.
The schematic diagram is below:
I do appreciate for any help!!!
That is a misconception. The molecule ID can be chosen completely free and does not have to have a relation to your actual molecules.
But please note the following quote from the fix rigid documentation.
For bodystyle molecule, atoms are grouped into rigid bodies by their respective molecule IDs: each set of atoms in the fix group with the same molecule ID is treated as a different rigid body. This option is allowed for both the rigid and rigid/small styles. Note that atoms with a molecule ID = 0 will be treated as a single rigid body. For a system with atomic solvent (typically this is atoms with molecule ID = 0) surrounding rigid bodies, this may not be what you want. Thus you should be careful to use a fix group that only includes atoms you want to be part of rigid bodies.
So you can create one group that has only the atoms you want to keep rigid and a second group that has only the non-rigid atoms and then use the molecule ID to define the different rigid bodies and do time integration with fix nve or similar using the second group for the non-rigid atoms.
It is really just a matter of properly studying the documentation. 
Thank you! It really helps !!!