From the top of my head, I would tell you to (1) check if you have correctly built the connectivity of your system, (2) check if you correctly input the parameters of your potentials, (3) check if your initial configuration is suitable for starting an MD simulation (maybe there are atoms sitting very close to each other) and (4) check the timestep value you have input.
Thank you, I’ve got the result! It didn’t work under the Linux system. I changed to windows and it worked. It could be a communication issue between systems.
hmmmmmm…

I dont think this should be a solution to that problem… I dont understand much about machines but I guess you just changed the specifics of who is doing the math for you and possibly the amount of parallel processes you have. The simulation will not be precisely the same (i.e., microstates attained after each timestep will not be the same) but whoever is the culprit of your “missing atoms” errors is still there and probably you are going to get the simulation to crash again at some point in windows.
More likely due to using a different number of processors when running in parallel (or not in parallel).
This doesn’t solve the problem but just hides it.
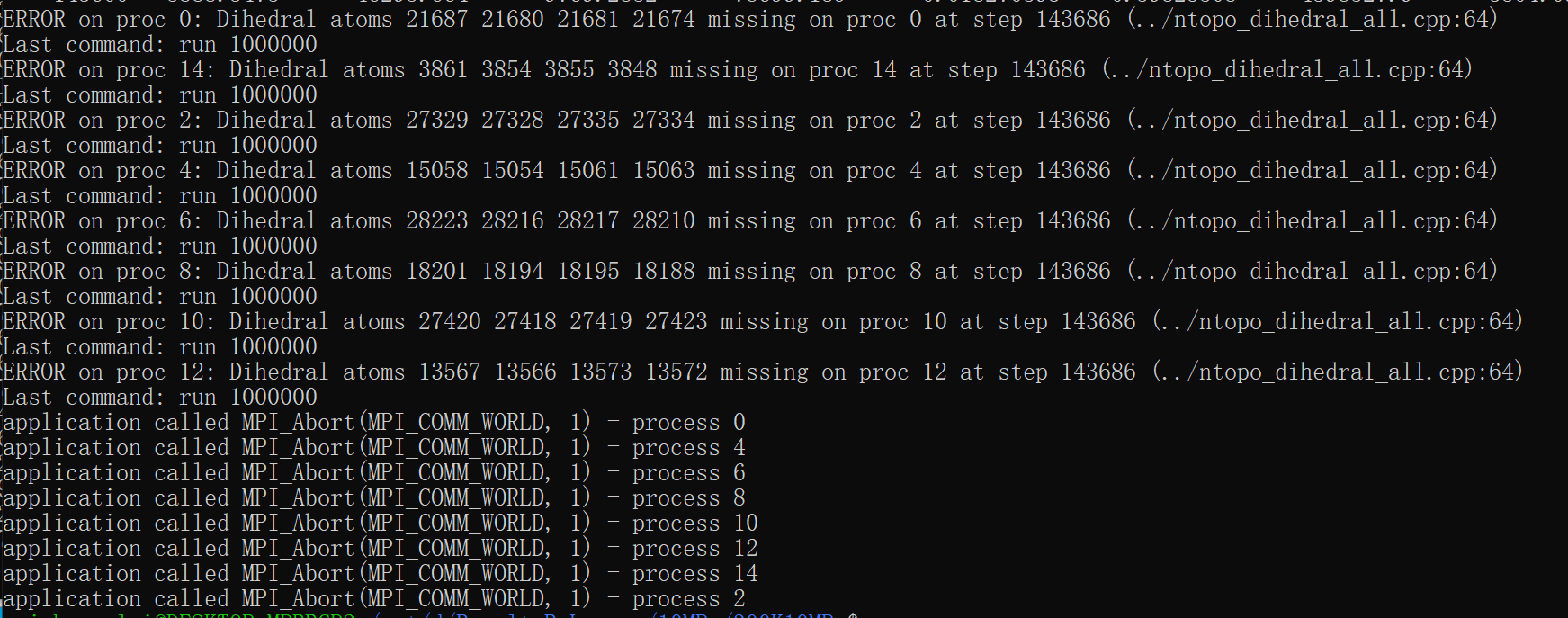
There are two main reasons for getting the “Dihedral atoms missing” error:
- your communication cutoff is too small due to using a pair style with a cutoff too short for including a fully stretched dihedral. LAMMPS should print a warning in that case
- atoms are moving very fast (or the simulation box is changing very fast) so they pass through the whole ghost atom region between neighbor list updates. This can have multiple reasons like very high potential energy of the initial geometry (remedy: run a minimization), or bad potential parameters (remedy: find a more suitable force field), or bad simulation parameters like too large a timestep or too infrequent neighborlist updates (remedy: use more conservative settings)
I only changed the number of running steps. The first time I planned to run 1,000,000 steps at a time, and then it failed at 140,000 steps. So I divided 1,000,000 into 100,000 steps and finally succeeded. I feel weird too, but I don’t have a better solution.
Thank you!I’ll give it a try when I get a chance
If you truly believe you solved the underlying issue either by changing the machine or by splitting your simulation into 10 parts, all I have left to say is that I will wear black tomorrow to mourn for the mental stability of the person supervising you