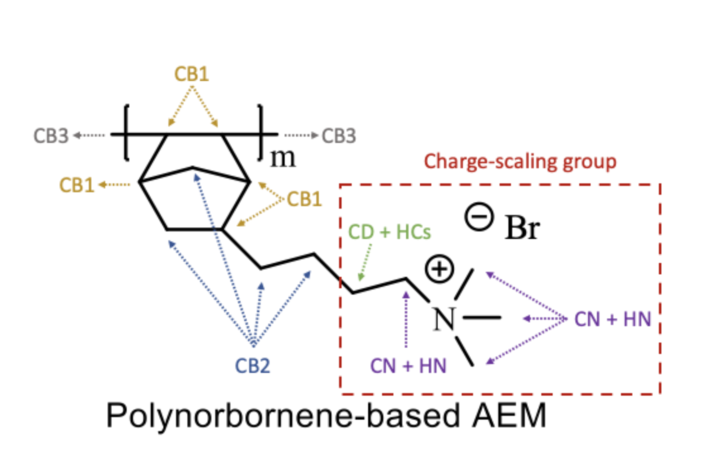
I’ve been using Enhanced Monte Carlo (EMC) to create polymer membranes system as an input for LAMMPS , and up until now, the process has been rather simple. I have successfully created Poly norbornene system with butyl quaternary ammonium as it’s side chain. the system also has equal number of Hydroxide ions as the quaternary ammoniums to neutralize the system .It has certain amount of water molecules as well. However the resultant polymer.data file does not have different atom types for the carbons in the linking site with other monomers, in norbornene ring, the carbons in butyl side chain and the carbons in quaternary ammonium. Whereas in literature the difference in these type carbons has been reported. Even the partial charges for the atom types in the EMC output file is different from what has been reported.
the following is the EMC .esh script that I have been using, where it generates polymer chain with 2 monomers, 2 hydroxide ions and 6 water molecules
#!/usr/bin/env emc.pl
#
# OPTIONS
# -------------------------
ITEM OPTIONS
field opls/2024/opls-aa
number true
density 0.95
ewald true
replace true
pdb_licorice true
emc_execute true
ITEM END
ITEM GROUPS
# monomer with built-in [N+] is good
monomer C1(C(C2C(CC1C2)CCCC[N+](C)(C)C)(*))(*), 1, monomer:2
terminator *C, 1, monomer:1, 1, monomer:2
OH_ion [O-]
# Prefer an OPLS water template if accessible; if not, keep this and set charges in LAMMPS
Water O
ITEM END
ITEM CLUSTERS
polymer random, 1
OH_ion OH_ion, 2
Water Water, 6
ITEM END
ITEM POLYMERS
polymer
2 monomer, 2, terminator, 2
ITEM END
the above script generated polymer.data which can be used as input for LAMMPS
LAMMPS output created by EMC v9.4.4, build Jul 31 2025 20:33:49
112 atoms
107 bonds
198 angles
315 dihedrals
7 atom types
5 bond types
7 angle types
7 dihedral types
0 10.09696439 xlo xhi
0 10.09696439 ylo yhi
0 10.09696439 zlo zhi
Masses
1 12.01100 # c4
2 1.00790 # h1
3 1.00790 # h1o
4 1.00790 # h1w
5 14.00670 # n4
6 15.99940 # o1h-
7 15.99940 # o2w
Pair Coeffs
1 0.06600 3.50000 # c4,c4
2 0.03000 2.50000 # h1,h1
3 0.00000 2.50000 # h1o,h1o
4 0.00000 2.50000 # h1w,h1w
5 0.17000 3.25000 # n4,n4
6 0.25000 3.20000 # o1h-,o1h-
7 0.15540 3.16557 # o2w,o2w
Bond Coeffs
1 268.00000 1.52900 # c4,c4
2 340.00000 1.09000 # c4,h1
3 367.00000 1.47100 # c4,n4
4 553.00000 0.94500 # h1o,o1h-
5 600.00000 0.95720 # h1w,o2w
Angle Coeffs
1 58.35000 112.70000 # c4,c4,c4
2 37.50000 110.70000 # c4,c4,h1
3 80.00000 111.20000 # c4,c4,n4
4 50.00000 113.00000 # c4,n4,c4
5 33.00000 107.80000 # h1,c4,h1
6 35.00000 109.50000 # h1,c4,n4
7 75.00000 109.50000 # h1w,o2w,h1w
Dihedral Coeffs
1 0.70000 0.35000 0.05000 0.40000 0.00000 # c4,c4,c4,c4
2 0.15000 -0.45000 0.00000 0.60000 0.00000 # c4,c4,c4,h1
3 1.37950 0.63850 0.22900 0.97000 0.00000 # c4,c4,c4,n4
4 0.72710 0.32310 0.12380 0.52780 0.00000 # c4,c4,n4,c4
5 0.15085 -0.45255 0.00000 0.60340 0.00000 # c4,n4,c4,h1
6 0.15000 -0.45000 0.00000 0.60000 0.00000 # h1,c4,c4,h1
7 0.19200 -0.57600 0.00000 0.76800 0.00000 # h1,c4,c4,n4
The attached is the schematic diagram of the poly norbornene system with different carbon atom types
And also to test the EMC .esh script I have generated the Poly styrene system as well, due to the presence of aromatic ring there are different carbon atom types. This shows the EMC ability to differentiate the carbon atom types.
Ultimately I need to generate a polymer system with different carbon atoms for the linkers,for the carbons in the ring, in the side chain and finally in the quaternary ammonium. Please guide me for building such system