I am unable to maintain vacuum in fix ttm/mod electron grids.As per documentation,
Visual representation of the simulation domain
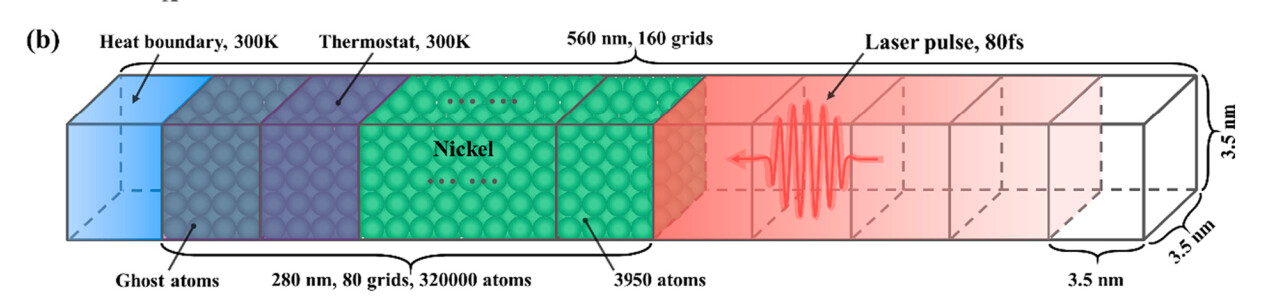
So there will be 160 grids, but 80 of them in vacuum
I defined the dimensions of simulation domain like following and populated it with atoms
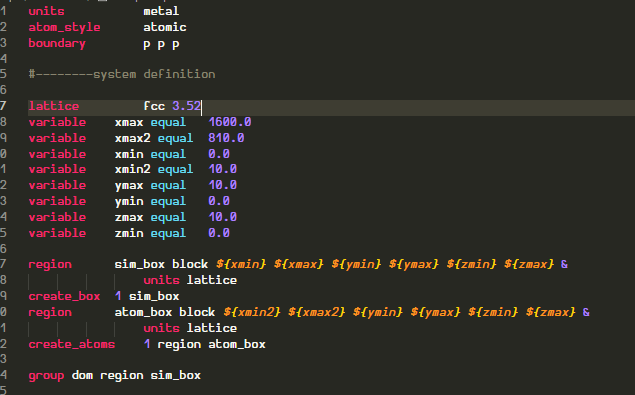
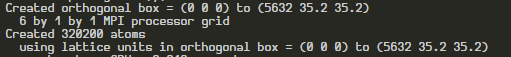
I am getting nearly correct number of atoms
Defined fix ttm/mod like the following
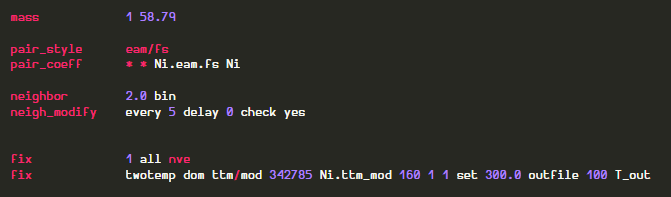
A fix accepts group-ID. So instead of ‘all’ I made the ‘dom’ group to inclued the empty space in simulation box.
this is the configuration of the init file (vacuum boundaries)
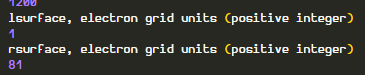
Index starts from 0 in electron grids. The 1st block is empty so it start from 1 and plus 80 = 81.
Unexpectedly, the output file contains electron temperature in all grids (after 81 it should be zero)

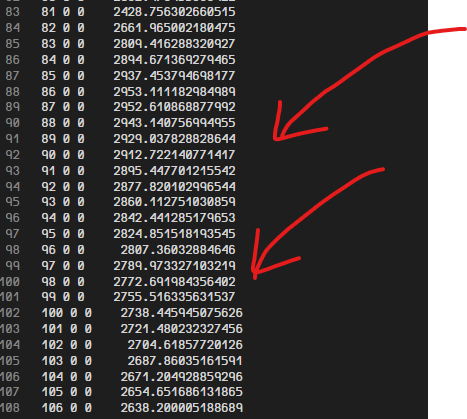
I alreay tried fix ttm/mod applied to ‘all’ group. Same issue. I thought maybe the grid division was only happening inside the atom filled region. So, I tried to include the empty space with ‘dom’ group. Also used infile command to initialize electron temperatures where temperature is zero outside of atom filled region.
But on the other hand when I Check the lattice temperature with computes(fraction = 1/80)
Half the grid has zero temperature(after 80)
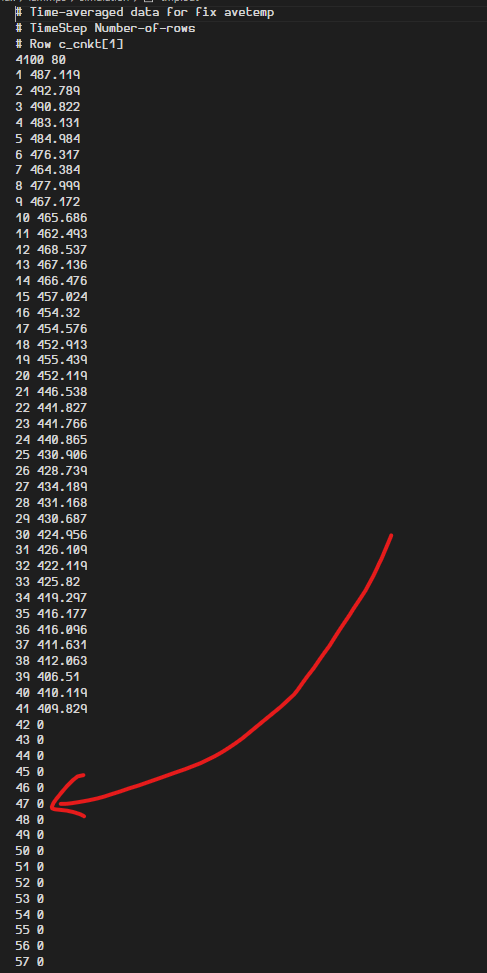
How to make the empty grids have zero temperature (DEFINE VACUUM) ?
why is half the lattice getting frozen?
And in fix ttm/mod in which surface does the heat flux hits?? I am confused. I needed it to be on the r_surface. But temperatue is high on the left end. Do I need to invert the x dimensions then?
Any suggestion would be appreciated. Thank you