Dear All,
I am trying to calculate the surface tension of 1024 Tip4P/2005 water molecules at 300K and 1 bar using lammps.
The box lengths are ly=lz=31 A and lx=93.1 A (largest dimension). I ran an NVT over this system with temperature set to 300K. However, the computed normal pressure from lammps is (pxx in this case) -12.4 atm. But I want it to be close to 1 bar so that I can make comparisons with experimental results.
Running an NPT over this system changes the box dimensions, which I do not want because I am using the equation st = 0.5 * lx*<Pxx-0.5*(Pyy+Pzz)> to evaluate the surface tension.
Also, If I first try to run an NPT, then switching from NPT to NVT gives the error “bond extent greater that half the periodic box length.”
How can I ensure system pressure close to 1 atm.
Sincerely,
Neha, India
How about running NPT after your NVT run to have the pressure 1 atm. Then switch back to NVT (with new dimensions) to calculate surface tension…
Dear All,
I am trying to calculate the surface tension of 1024 Tip4P/2005 water
molecules at 300K and 1 bar using lammps.
The box lengths are ly=lz=31 A and lx=93.1 A (largest dimension). I ran an
NVT over this system with temperature set to 300K. However, the computed
normal pressure from lammps is (pxx in this case) -12.4 atm. But I want it
to be close to 1 bar so that I can make comparisons with experimental
results.
Running an NPT over this system changes the box dimensions, which I do not
want because I am using the equation st = 0.5 * lx*<Pxx-0.5*(Pyy+Pzz)> to
evaluate the surface tension.
Also, If I first try to run an NPT, then switching from NPT to NVT gives the
error "bond extent greater that half the periodic box length."
there is a lot of crucial detail missing in your description to give
dependable advice.
for example, how can we know that you did what you say you did correctly.
it is quite common to receive posts like yours only to find out that
there were issues with the system set up or the input script and that
the script actually didn't do with the person thought it would, or
that there were inadequate choices made to the problem at hand.
axel.
More physics info would also help. Do you have some sort of interface in your system? Most folks on this list aren’t familiar with Tip4P. Are there publications using Tip4P that show P = 1 atmosphere at 300K in systems comparable to yours? If not, why do you think P should be 1atm for your geometry at T = 300K? There’s more than one way to reduce the pressure of a liquid-vapor system. Obviously you need to pick the one that corresponds to your physics problem, but you didn’t give us enough information for us to give the best advice.
Rob
I am sorry for incomplete information in the first mail.
I am trying to simulate a VLE system for 1024 Tip4p2005 water molecules which has two interfaces.
I am referring to the paper http://dx.doi.org/10.1063/1.2715577 . They have simulated 1024 Tip4p2005 water. Their simulated normal pressure is -0.02 bar at 350K.
ly and lx in their system is 30 A ( 31 A in my system)
lz in their’s is 100 A (mine 93 A)
I am attaching my input script and the data file I have used.
Also, I am not able to switch from NPT to NVT ( with changed dimensions) as I have mentioned in my earlier mail.
input.vap_liq.txt (2 KB)
water1024.txt (229 KB)
this input deck has several errors and doesn't work as it is.
your claim about not being able to use fix nvt doesn't make sense either:
fix npt and fix nvt are essentially the same code with fix npt doing
one extra step on top of fix nvt.
using fix npt for this kind of system makes little sense. fix nvt
should work just as well. with the free surface, the system should
relax into its "native", no-pressure structure, unless you configure
fix npt to use isotropic scaling of all dimensions, which is plain
wrong. if you do want to maintain the shape in x and y-direction, you
can set up fix npt to couple those two directions and leave z alone.
yet, fix nvt should be just fine and avoids many complications.
axel.
In a separate run, I had run fix nvt on the system for 5 nanoseconds (no npt) , and at the end of which the normal pressure was -12 bar. It did not relax into its “native”, no pressure structure of approx 1 bar normal.
In a separate run, I had run fix nvt on the system for 5 nanoseconds (no
npt) , and at the end of which the normal pressure was -12 bar. It did not
relax into its "native", no pressure structure of approx 1 bar normal.
where do you get the information from that it should be 1 bar for TIP4P/2005?
but if you run like in this input a tip4p coulomb style with plain
ewald (which LAMMPS doesn't allow without modifications of the source,
and for a good reason), your simulations will be bogus regardless.
upon visualization, your system doesn't look like a slab configuration
either. this looks like a broken simulation all along.
axel.
I am referring to the paper http://dx.doi.org/10.1063/1.2715577 . They have simulated 1024 TIP4P/2005 water molecules, ewald sums to deal with electrostatic interactions and report normal pressure of -0.02 bar at 350 K.
My system looks like below at the end of 5 ns run on lammps-23Sep13.
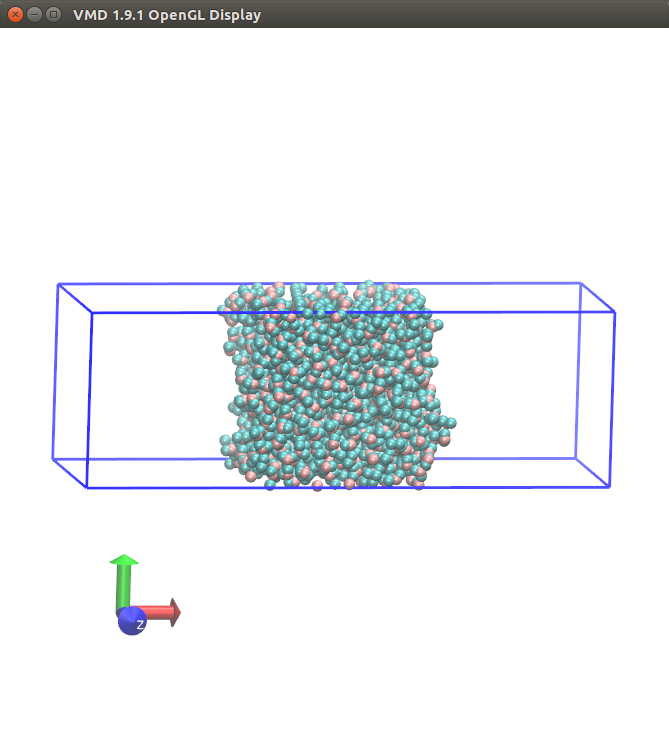
I am referring to the paper Surface tension of the most popular models of water by using the test-area simulation method | The Journal of Chemical Physics | AIP Publishing . They
have simulated 1024 TIP4P/2005 water molecules, ewald sums to deal with
electrostatic interactions and report normal pressure of -0.02 bar at 350
K.
[...]
just to mention, I have another simulation on TIP4P/2005 water model, which
uses
kspace style* pppm/disp/tip4p and *
*kspace_modify force/disp/real 0.0001*
*kspace_modify force/disp/kspace 0.002*
if you want to compare to the publication from above, you probably need to
use pppm/tip4p, even though pppm/disp/tip4p is probably more correct for a
slab configuration.
*However, the surface tension values which I am getting for this is lower
(64.97 mN/m , STDEV 1.18) when compared to the experimental value . Using
ewald gives surface tension (67.68 mN/m STDEV 1.025) value closer to the
experimental value .*
whether you can reproduce experimental values is a concern only, if your
simulation as such is correct. ever heard of the term "error
compensation"? what is the value of reproducing an experiment for the wrong
reason? typically, while you may have a better match for one observable,
others will be worse.
Sir, can you tell me why using ewald with tip4p couloumb style is a bad
idea?
because with kspace style ewald, the position of the negative charge is
wrong. with a TIP4P pair style, the point M for the negative charge is not
explicitly defined but rather computed on the fly. pppm/tip4p takes care of
this, ewald doesn't. thus you compute in the kspace part a TIP3P geometry
with TIP4P charges, which is flat out wrong. as i mentioned before, current
LAMMPS versions detect this and abort with an error message (as they
should).
axel.
sir, if I have to compare my result with an experiment which determines the surface tension of water VLE system at atmospheric pressure, then how do I ensure same normal pressure in the simulation.
-Neha