Hello,
I’ve been querying the database, and I’m getting a lot of structures such as mp-1184151 which are not well converged. In this particular case, the energy is probably much too low, since this isn’t the experimental ground state of Cs, and the band gap is non-zero. What’s the best way to exclude structures like this from my query?
I can’t find the API equivalent for the " 1. Large change in a lattice parameter during relaxation.
2. Large change in volume during relaxation." tags, or the “gap value is approximate and using a loose k-point mesh”. The best work-around I have right now is to get the list of task_ids and exclude anything where that list only contains one element.
More broadly, I think structures like this one are going to cause problems for phase diagram generation, since the energy difference is so large.
{"warnings": {
"$nin": [
"Large change in a lattice parameter during relaxation.",
"Large change in volume during relaxation."
]
}}
to exclude materials with particular warnings with respect to the structure. The gap value is approximated when a full bandstructure has not been calculated for a material. For API queries, the list of calculations that a material has does not include “bandstructure”, i.e.
{"has": {"$nin": ["bandstructure"]}}
You can merge the above criteria in e.g. a call to pymatgen’s MPRester.query method (dispatching to our /rest/v2/query API endpoint).
We understand your concern and interest in constructing phase diagrams using a selected subset of our materials. I hope this info helps you going forward in the near term.
What criteria are you using to determine these are badly converged? Note that having a band gap from a calculation not targeting electronic structure, doesn’t mean much. Our structure optimizations are trying to get the right structure and formation energy in high throughput. We run a separate set of calculations to determine true band gap.
Also, how are you building phase diagrams without any elemental entries?
The parameters for relaxations are coarse, and I didn’t think they were good enough to give accurate energies for convex hulls. I had thought that Materials Project convex hull energies were calculated from static runs for this reason, but after some exploring, it seems that they’re actually done with energies from relaxations.
I decided to test the convergence by running a StaticFW for the suspect Cs structure, mp-1184151, but I couldn’t converge it electronically. I noticed that even with the increased k-point density of the StaticFW, the k-point generator still only assigned a gamma-point only calculation for this cell, so I increased the k-point grid to 3 x 3 x 3. This calculation returned an energy (E0 in the OSZICAR) of -24.471127 eV, much higher than the relaxation, which gave -25.966. Therefore it seems that the single k-point is inadequate to sample this cell, even approximately. I also tested running an OptimizeFW on mp-1184151, and it timed out after around 30 ionic steps, suggesting that the single gamma point is also inadequate for relaxing this cell.
In this particular case it seems that it’s not a difference between default OptimizeFW and default StaticFW; instead, they both fail for the large Cs cell. However, in general I would prefer to make convex hulls using energies from static calculations, and ignore phases which don’t have static calculations yet. For example, I noticed that going by relaxations, the mp-21462 structure of Eu is 19 meV/atom more stable than mp-623532, but according to the more accurate static calculation, it is 2 meV/atom less stable. I should be able to exclude relaxation-only phases by using the filters @dwinston described.
Thanks for the help!
P.S. While we’re talking about polymorphs of Cs, do you know why this relaxation stopped after three steps? https://materialsproject.org/tasks/mp-639727#mp-639727 The energy change between the second and third steps is 28 meV, about an order of magnitude larger than EDIFFG for this system.
Ah! That digging helps a ton. I’ll look into if we need to rerun Cs with higher k-points which suggests that the structures might also change by a small amount. Can you elaborate on what you mean by “timed out after 30 ionic steps”?
While the static calculations are indeed at a higher k-point density, I wouldn’t take higher k-points as any finer or coarser. K-point convergence isn’t monotonic, so it depends on what your goal is. For compositional thermodynamics it is more important to have a consistent parameter set in which you’re in approximately the same place on this convergence manifold across all the constituent entries. In your example, you can’t just assume that going from gamma-point to 3 x 3 x 3 is monotonic. It’s also a question of what density you’d use for the other calculations and being consistent.
I would still be wary of using the static’s for the phase diagram construction over the structure optimizations. There’s a lot more eyes looking at the structure optimization energies for various applications, where as the static calculations are run with the intent of extracting band structure.
This specific structure is not related to that issue. That issue is really related to a legacy problem in running calculations where if the volume changes by too much the k-points don’t get updated accordingly. We built the deprecation for that, but we’re also considering deprecating more calculations as we have a large legacy set from ~ 2012,2013 and VASP itself has changed quite a bit since then. So, what is a “complete” calculation today, might not be one tomorrow as we get better and better at running these in an automated fashion.
As for the Cs task (mp-639727), I’m not sure. That is one of the old 2013 calculations and despite how popular VASP is, it still baffles me on a regular basis. I’d completely expect it to have respected the EDIFFG of 0.002, but it looks like it didn’t.
I ran the relaxation in the development queue, and it hit maximum walltime after 30 ionic steps.
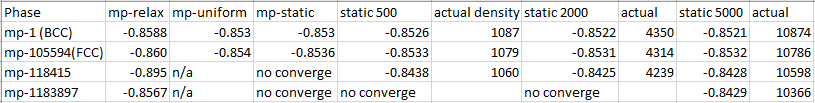
I’ve done some convergence testing for the four most stable polymorphs of Cs in the Materials Project, by increasing the k-point density in the MPStaticSet to 500, 2000, and 5000. Here are the energies/atom in eV.
It looks like the two 1-atom primitive cells (FCC and BCC) can be safely compared to each other at any equal level of convergence (the energy difference converges quickly, that is). The 20-atom mp-1183897 has less error cancellation, but still not terrible, and it’s just the 29-atom mp-118415 which has an unreasonably low energy in the relaxation.