Dear developers :
I am working on an interatomic potential for ZrC. This potential was implemented in the MD program developed by Ju Li’s group, however it seems that a counterpart in LAMMPS is still lacking.
The potential generally follows the EAM formulae (more specifically, in the form proposed by Willaime and Massobrio) but with a critical difference : when considering the binding energy between a pair of Zr and C , a screening function should be multiplied (i.e. the Equation 16 and 17 in the original paper) :

where the illustration of angle theta can also be found in Fig.3 in the original paper :
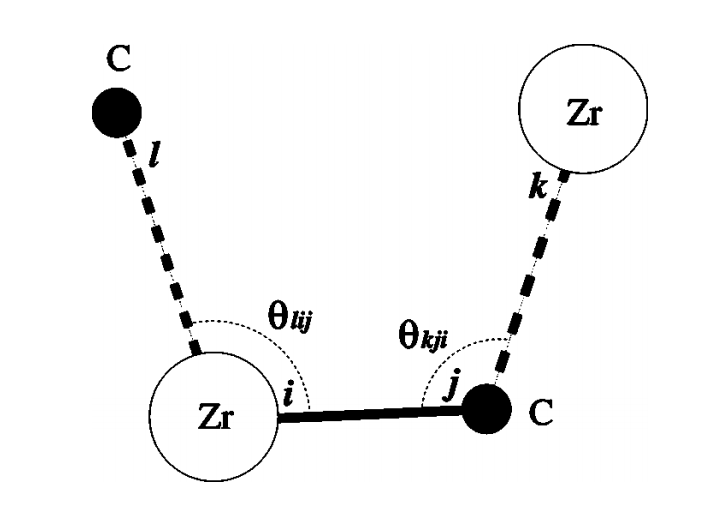
The tricky part, in my opinion, is exactly the implementation of such an “angular-dependent” screening function. I’m not sure if LAMMPS currently supports such an EAM, and I think a new pair_style might be needed to implement it. According to the book Extending and Modifying LAMMPS : Writing Your Own Source Code, it’s better to find a similar pair_style and modify it. But I didn’t find more useful information. Could anyone please shed some light on the the angular-dependent screen function? (For example, is there a many-body potential in LAMMPS that has similar screening functions?)
Any comments and suggestions would be appreciated, thanks!