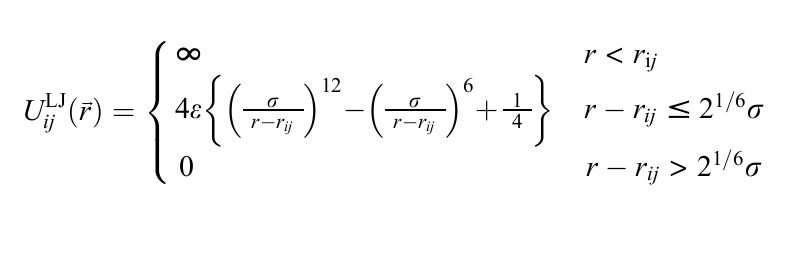Dear lammps users,
I am using lammps to calculate the coarse-grained polymer system via LJ nonbond potential table of my own. The function of the LJ bonbond potential is like this:
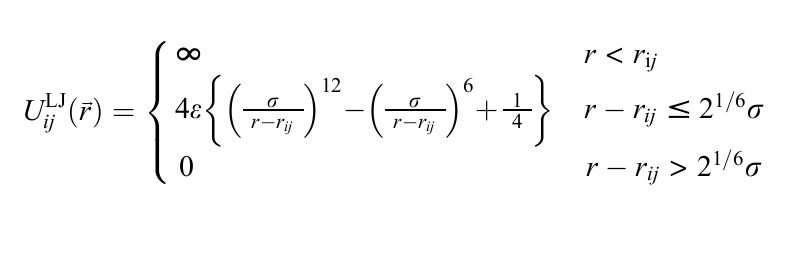
In the piecewise function, the first part from zero to rij is an infinite range. My question is whether the lammps could express the infinite energy in the nonbond potential table.
Best Regards
D. Chai
Dear lammps users,
I am using lammps to calculate the coarse-grained polymer system
via LJ nonbond potential table of my own. The function of the LJ bonbond
potential is like this:
In the piecewise function, the first part from zero to rij is an infinite
range. My question is whether the lammps could express the infinite energy
in the nonbond potential table.
this is not needed (and not meaningful). just define your tabulation
between a reasonable inner cutoff which is larger than your r_ij. due to
the shape of the potential, particles must never come close this inner
cutoff anyway.
... or you can just use the lj/expand pair style.
axel.

Thanks a lot, Axel. The important rules have been paid attention to, but my calculation model (in the attachment) always have some atoms which run close to the inner cutoff after 50000 steps (less then 5 minutes calculation with 4 cores), and the error “Bond atoms 156 157 missing on proc 1 at step 53290” arises. I think that’s because the the atoms is thrown far away by the abrupt big force near the inner cutoff. what should I do in lammps to prevent from the error.
Best regards
D. Chai
LJ_tablepotential.tar.gz (105 KB)
Thanks a lot, Axel. The important rules have been paid attention to, but
my calculation model (in the attachment) always have some atoms which run
close to the inner cutoff after 50000 steps (less then 5 minutes
calculation with 4 cores), and the error "Bond atoms 156 157 missing on
proc 1 at step 53290" arises. I think that's because the the atoms is
thrown far away by the abrupt big force near the inner cutoff. what should
I do in lammps to prevent from the error.
this may not be a direct consequence of the potential, but rather
something indirect, e.g. too large a time step or too infrequent update of
the neighbor lists or too short a communication cutoff, or just a badly
conditioned configuration. i doubt that with a change of the potential
alone this can be solved.
axel.
Dear Axel,
You are right. My mistake takes place due probably both to too large time step and to the infrequent update of the neighbor lists. Thanks for the key hint.
Best wishes
D. Chai