Dear all,
I’m trapped in the calculation of region temperature. The version of 20160216 lammps is employed. Two methods are used in my simulation about inorganic aqueous solution. One is by the command of compute temp/region and the other is by the command of compute ke/atom. The specific procedures are as follows:
Both methods give the consistent temperatures in hot or cold region. But it is very confusing that the calculated temperature is wrong. For example, if the system is kept at 300K by Nose-Hoover thermostat, the region temperature will be about 212K, which is quite unreasonable.
In order to find out what should account for the wrong region temperature, I simulated pure SPC/E water maintained at 300K by ose-Hoover thermostat under NVT ensemble. Two conditions are simulated. A part of the log file is showed :
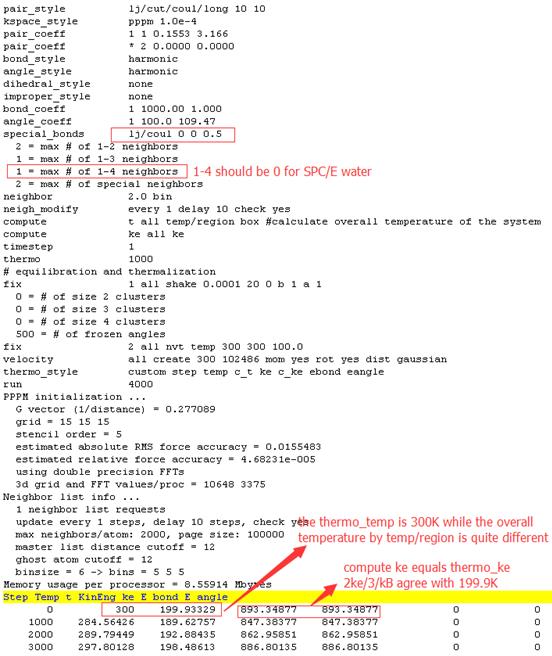
Results of the first condition using special_bonds lj/coul 0 0 0.5
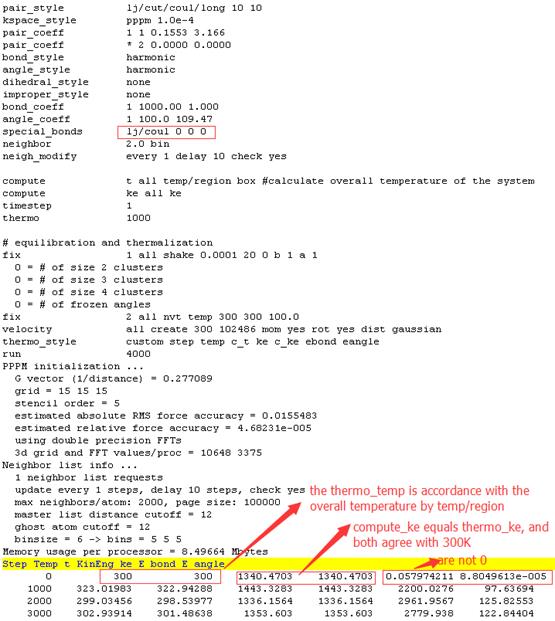
Results of the second condition using default special_bonds lj/coul 0 0 0
From results above, mainly two problems exist.
Firstly, from http://lammps.sandia.gov/doc/compute_ke.html, thermo_ke corresponds to thermo_temp and is different from compute_ke. But in the first condition, thermo_ke doesn’t agree with thermo_temp. For both conditions, thermo_ke equals compute_ke completely.
Secondly, from http://lammps.sandia.gov/doc/compute_temp_region.html, compute temp/region differs from other compute temp because it does not subtract out degrees-of-freedom due to fixes that constrain motion, such as fix shake. In the first condition, temp/region and ke give wrong overall temperature and ebond as well as eangle equals 0. In the second condition, temp/region and ke give right overall temperature but ebond and eangle are not 0. So I doubted whether both ebond and eangle should be 0 when fix shake is used to constrain bonds and angles. The only difference of inputfile in two conditions above is the special_bonds settings. In the first condition, I think the auto-generation special neigbors in lammps is wrong. From auto-generation in logfile, when using lj/coul 0 0 0.5, 1-4 neigbors are 1 for water but it is 0 actually.
So, now, I have no idea how to get the right region temperature under the condition when fix shake is used, ebond and eangle are always 0 regardless of any special_bonds settings,which happens in my aqueous solution simultion.
Can anyone tell me how to solve the problem? Thank you very much for attention.
Best regards,
Tingting Chen