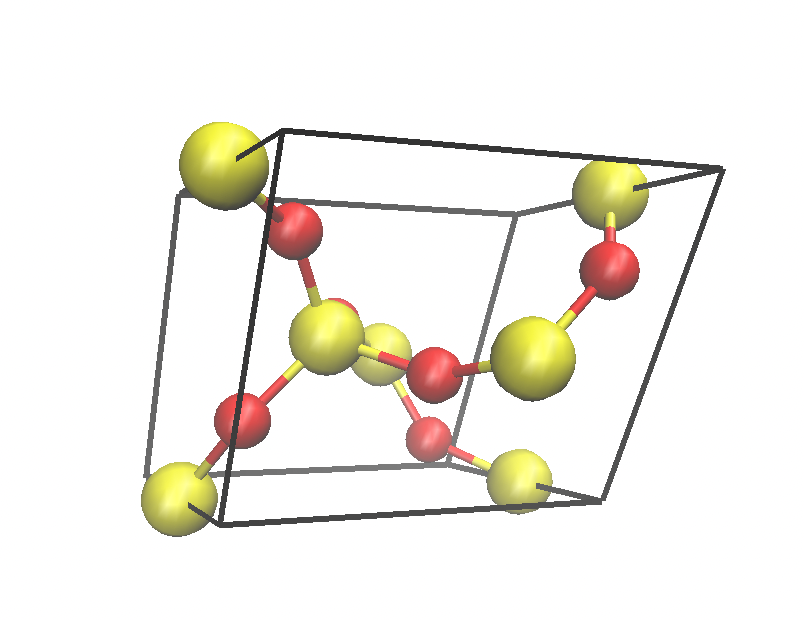
I download the Quartz cell here. Quartz crystal structure
I downloaded the *.cif file and opened it with VMD. I found that the structure looked terrible (Fig. 1). Then I opened *.cif file with VAST (see Figure 2). I deleted the atoms outside the cell box and finally got the structure in Figure 3. Then I used Matlab to write the structure in xyz format as a data file. Then I used replicate to get the bigger structures.
I checked the structure after MD simulation with reaxff. It looks very terrible. Check the out file, I found that when I used replicate, the number of atoms in the system increased, and the tilt also increased exponentially, making the structure of MD simulation very strange.
two questions:
(1) This is my first time to run crystal structure with 90 90 120 degree, my initial crystal cell structure is right?
13
Si O2
Si 2.309045 0.000000 0.000000
Si 2.309045 0.000000 5.405400
Si -0.148955 4.257381 0.000000
Si -0.148955 4.257381 5.405400
Si -1.154523 1.999692 3.603600
Si 3.761477 1.999692 3.603600
Si 1.303477 2.257689 1.801800
O 1.376726 1.136295 0.643783
O 0.295697 1.760427 2.959817
O 3.243577 0.624132 4.247383
O 0.785577 3.633249 1.158017
O 2.753697 2.496954 2.445583
O -1.081274 3.121086 4.761617
(2) triclinic simulation with replicate, the tilt is also changed. How to run it?
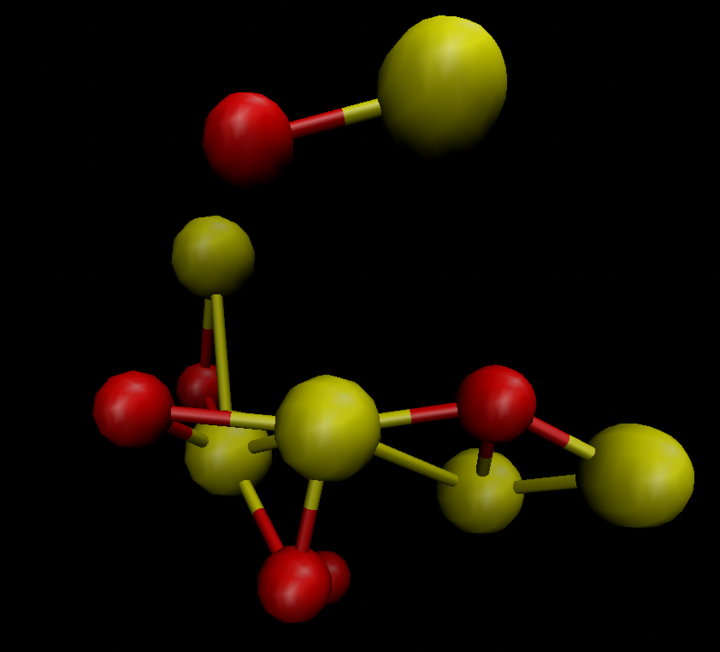
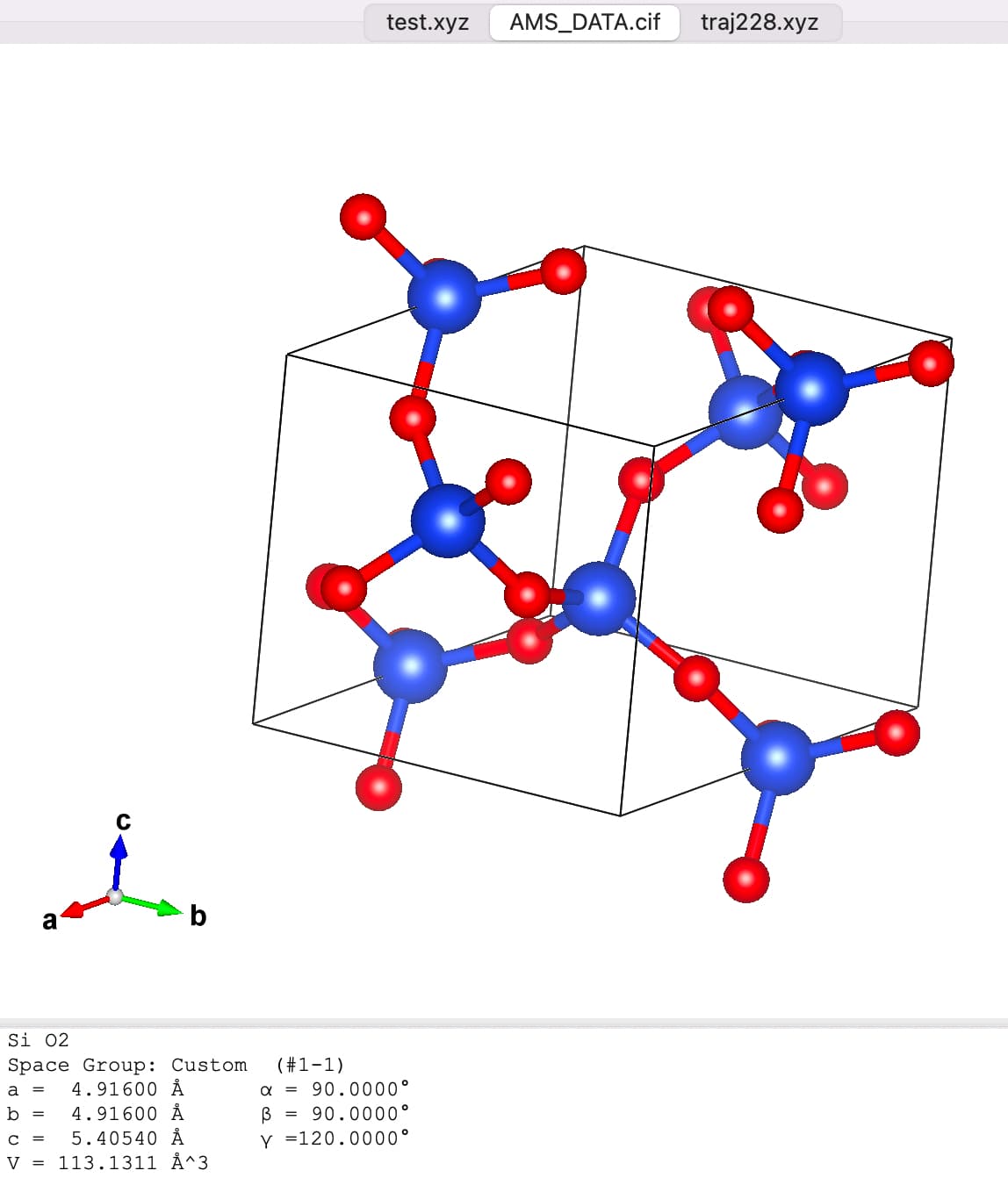
input file and out file are as follows:
preequi.inp
#-------------------------- Initial Setup -------------------------------------#
units real
atom_style charge
dimension 3
boundary p p p
timestep 1
#------------------------------------------------------------------------------#
read_data Quartz_AMS_DATA_vmd.data
replicate 2 2 4
Quartz_AMS_DATA_vmd.data
13 atoms
0 bonds
0 angles
0 dihedrals
0 impropers
2 atom types
0.00 4.9160 xlo xhi
0.00 4.9160 ylo yhi
0.00 5.4054 zlo zhi
5.0 0.0 0.0 xy xz yz
Masses
1 16.00
2 28.09
Atoms
1 2 2.40 2.31 0.00 0.00
2 2 2.40 2.31 0.00 5.41
3 2 2.40 -0.15 4.26 0.00
4 2 2.40 -0.15 4.26 5.41
5 2 2.40 -1.15 2.00 3.60
6 2 2.40 3.76 2.00 3.60
7 2 2.40 1.30 2.26 1.80
8 1 -1.20 1.38 1.14 0.64
9 1 -1.20 0.30 1.76 2.96
10 1 -1.20 3.24 0.62 4.25
11 1 -1.20 0.79 3.63 1.16
12 1 -1.20 2.75 2.50 2.45
13 1 -1.20 -1.08 3.12 4.76
output