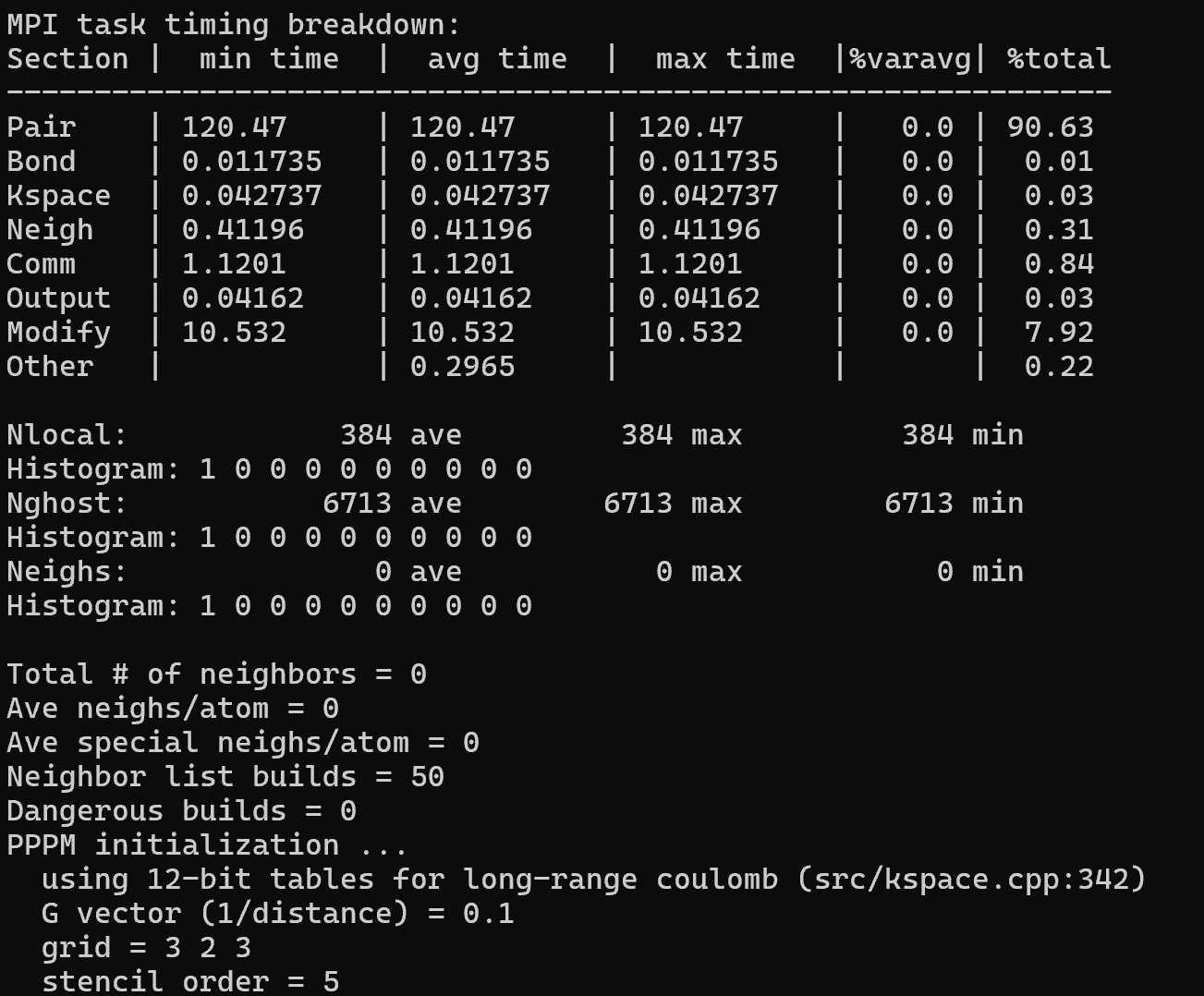
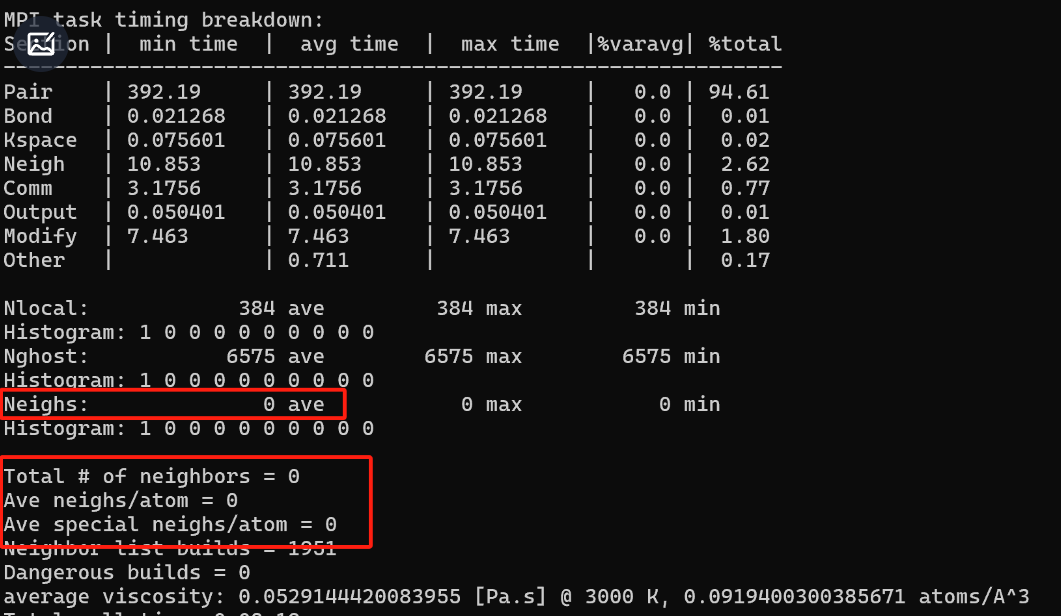
I am using LAMMPS to calculate viscosity and can obtain the result, but I found that neights is 0 in the calculation process file. How can I modify this?Below are part of my codes:
units metal
dimension 3
boundary p p p
atom_style full
neighbor 2.0 bin
neigh_modify every 1 delay 0 check yes
variable T equal 3000 # run temperature
variable Tinit equal 3050.0 # equilibration temperature
variable V equal vol
variable P equal 1
variable dt equal 1 #dt
variable p equal 200 # correlation length
variable s equal 2 # sample interval
variable d equal $p*$s # dump interval
This is not properly formatted. Can you read it as it should? I cannot.
Please see see the forum guidelines post for how to properly format.
What is your concern here? Did LAMMPS not compute what you asked it to do?
Please note the neighborlist statistics is a pure diagnostic and with complex hybrid pair styles, LAMMPS may not find a representative neighbor list for the system. Even if it does, the results are for that one neighbor list only and not the remaining lists for other sub-styles.
Why do you use this setting? Your results can be seriously wrong, if you choose the wrong value. How did you determine this value? What does LAMMPS output for the PPPM setup, when you comment out this line and how does it differ from setting gewald manually?
In modeling there are no absolute “rights” or “wrongs”, but only choices that are better or worse for a specific purpose. Models are by definition always wrong, but they can still teach us important lessons.
Thank you for your reply. LAMMPS has provided results for the code I input, but I noticed that the boxed-out part shows 0, which is different from my previous simulations, so I had some doubts. Based on your response, can I understand that if LAMMPS can produce results, in the case where the pair_style is set to hybrid/overlay, there is no need to focus on the “neighs”?
I’ve already answered this. Let me quote it again:
Please note the neighborlist statistics is a pure diagnostic and with complex hybrid pair styles, LAMMPS may not find a representative neighbor list for the system. Even if it does, the results are for that one neighbor list only and not the remaining lists for other sub-styles.
In other words, the output you are looking is only meaningful when you have a single neighbor list covering all atoms. But with hybrid pair styles, this is usually not the case. If LAMMPS cannot find a suitable neighbor list, it will not output the neighbor list statistics.
If you worry about correctness of your results, you have to look at the forces though.