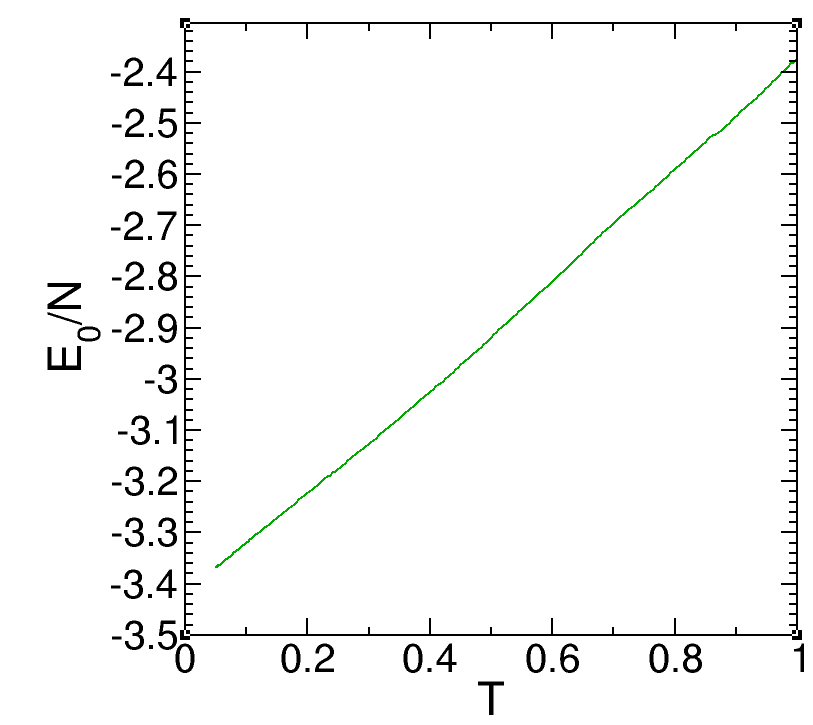
The above is the configuration details of the paper, and the following is the lammps script I wrote:
units lj
dimension 2
atom_style atomic
boundary p p p
timestep 0.005
region myreg block 0 40.8248 0 40.8248 -0.5 0.5
#egion myreg block 0 65.6062 0 65.6062 -0.5 0.5
create_box 2 myreg
create_atoms 1 random 1300 31235 myreg
create_atoms 2 random 700 53341 myreg
mass 1 1
mass 2 1
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0 2.5
pair_coeff 1 2 1.5 0.8 2.0
pair_coeff 2 2 0.5 0.88 2.2
#min_style cg
minimize 1.0e-4 1.0e-4 10000 10000 #
#The call terminal (shell) creates a folder to store the computed data
shell mkdir ./configs_log_e
shell mkdir ./configs_quench_e
shell mkdir ./configs_equ_e
log log_e.lammps
#generates an ensemble of velocities
velocity all create 4 34343 dist gaussian
reset_timestep 0
#sets parameters for the building of pairwise neighbor lists
neighbor 0.3 bin
neigh_modify every 1 delay 0 check yes
compute and thermo
compute peratom all pe/atom
compute pe all reduce sum c_peratom
compute 1 all temp/com
compute myTemp mobile temp/com
#compute_modify myTemp dynamic/dof yes
thermo 1000
thermo_style custom step atoms temp press pe c_pe ke etotal
thermo_modify flush yes
#--------melting at a high temperature---------
fix 1 all nvt temp 4 4 $(100*dt)
fix f2d1 all enforce2d
run 1000000
unfix 1
unfix f2d1
#-------------------quench---------------------
fix 2 all nvt temp 4 0.4 $(100*dt)
fix f2d2 all enforce2d
reset_timestep 0
dump 2 all custom 10000 ./configs_quench_e/heat.* id type xu yu x y
dump_modify 2 sort id
run 9000000
unfix 2
unfix f2d2
undump 2
#------------------equ----------------------
dump files at fixed intervals
variable f file merged_powers.txt
variable s equal “0+next(f)”
fix 3 all nvt temp 0.4 0.4 $(100*dt)
fix f2d3 all enforce2d
run 2000000
reset_timestep 0
dump 3 all custom 10000000 ./configs_log_e/heat.* id type xu yu x y
dump_modify 3 sort id
#dump_modify 3 format 3 .15lf format 4 .15lf #format 7 .15lf format 8 .15lf #Determine output accuracy
dump_modify 3 every v_s first yes
dump 4 all custom 10000 ./configs_equ_e/heat.* id type xu yu x y
dump_modify 4 sort id
run 5000000
unfix 3
undump 3
undump 4
write_data kalj_e.data nofix nocoeff
------------------------------end------------------------------
However, I found that the Potential energy per particle in the cooling process is inconsistent with the paper:
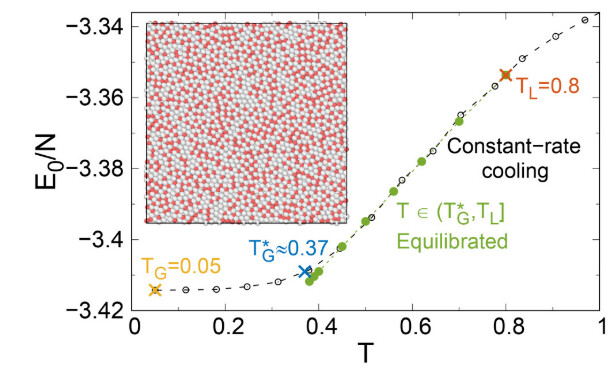
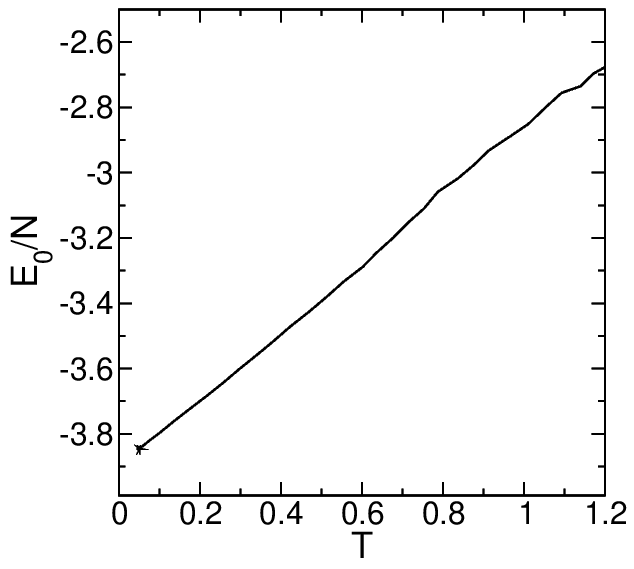
Did I ignore some commands in the process of writing the IN script, which caused the energy to deviate?I would be grateful if you could answer this question