I want to use ABOP potential to describe W-C system, and I found some papers provide the parameters like D0, r0, β and so on.
(“Analytical interatomic potential for modeling nonequilibrium processes in the W-C-H system” DOI:10.1063/1.2149492)
But I don’t understand how to use it because there is no specific command of ABOP in pair_style command, so I want to ask for assistance 
(1) Do I need a potential file (like EAM) to use the ABOP potential, or just need to write commands in input file (like L-J or Morse)?
(2) I learned that the ABOP potential comes from tersoff potential and found some clues in pair_style tersoff/table command, while I am confused.
Please give me some guidance, thank you a lot !!!
You should look up the original paper describing the ABOP potential. It should contain the information necessary to convert the ABOP parameters into the Tersoff parameters. Then you can use the converted parameters to generate a potential file for the tersoff pair style.
1 Like
Thank you so much !!!
The conversion of parameters between the paper and LAMMPS is very complicated (for me), but luckily I finally found a ABOP potential file for the W-C system on Github, the uploader’s id is houzf
I hope it can help someone who also needs this file
Thank you again 
Hey, it’s me again.
After getting the Tersoff potential file of WC, I tried to build a block of WC (as shown in the picture), but it could not keep the structure at 300K

I don’t understand whether the reason is that there is an error in the parameters or this potential file is not suitable for describing a stable WC structure
Also, I found that the papers that cite the original literature on W-C-H bond order potential all state that the BOP potential (not the Tersoff potential) was used to describe W-C. The BOP potential file is very long and complex (like EAM), perhaps you understand what means were used to generate the BOP potential file from the parameters? Thank u so much 
Are you sure that you are using the correct “units” settings? Your quoted input doesn’t show any units keyword and it is not likely that the parameter file was written for the default setting of reduced (aka “lj”) units.
Since you attached images instead of text files (why? it is so much more effort), it would be extremely laborious to make tests of our own and retype everything.
Sorry, you reminded me that I should have attached the file. I usually send screenshots when I ask people questions on social media apps,so I did it subconsciously (:з」∠)
But I found that new users can not upload attachments, so please forgive me for pasting the file directly 
[[[[[ in.WC ]]]]]
units metal
atom_style atomic
boundary p p p
neighbor 0.3 bin
#MODEL
lattice custom 2.900 &
a1 1.0 0.0 0.0 &
a2 0.0 1.732050808 0.0 &
a3 0.0 0.0 1.632993162 &
basis 0.0 0.0 0.0 &
basis 0.667 0.333 0.5
region box block 0 6 0 6 0 10 units lattice
create_box 2 box
create_atoms 2 box basis 1 1 basis 2 2
#MASS
mass 1 183.84 #W
mass 2 12.01 #C
write_data WC.lmp
#POTENTIAL
pair_style tersoff
pair_coeff * * WC.tersoff W C
#THERMO
thermo 1000
thermo_style custom step temp pe vol press
#300K, relax
velocity all create 300 6666 rot yes dist gaussian
fix 1 all npt temp 300 300 0.1 iso 0 0 1
dump 1 all custom 1000 WC_relax.lammpstrj id type x y z vx vy vz
timestep 0.001
run 50000
[[[[[[ WC.tersoff ]]]]]
#units:metal
#Ref.1 N. Juslin, P. Erhart, et al. Appl. Phys. 98, 123520, (2005)
# DOI: 10.1063/1.2149492
#Ref.2 M.V.G. Petisme, M.A.Gren, G. Wahnstr, Int. J. Refract. Hard Met, 49, 75-80(2015).
# DOI: 10.1016/j.ijrmhm.2014.07.037
#A,B = eV; lambda1, lambda2, lambda3 = 1/Angstroms; R,D = Angstroms, Other quantities are unitless
#Note:there is a typo for the "beta" value of C-C in Table 1 of Ref. 2, which is inconsistent with the one in Ref. 1.
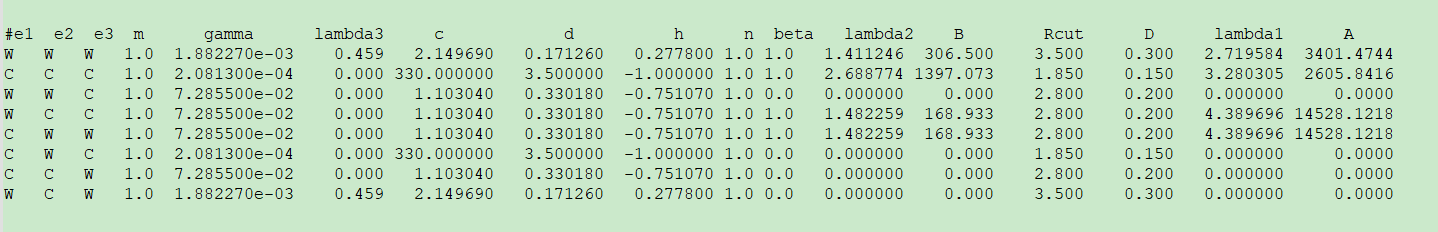
#e1 e2 e3 m gamma lambda3 c d h n beta lambda2 B Rcut D lambda1 A
W W W 1.0 1.882270e-03 0.459 2.149690 0.171260 0.277800 1.0 1.0 1.411246 306.500 3.500 0.300 2.719584 3401.4744
C C C 1.0 2.081300e-04 0.000 330.000000 3.500000 -1.000000 1.0 1.0 2.688774 1397.073 1.850 0.150 3.280305 2605.8416
W W C 1.0 7.285500e-02 0.000 1.103040 0.330180 -0.751070 1.0 0.0 0.000000 0.000 2.800 0.200 0.000000 0.0000
W C C 1.0 7.285500e-02 0.000 1.103040 0.330180 -0.751070 1.0 1.0 1.482259 168.933 2.800 0.200 4.389696 14528.1218
C W W 1.0 7.285500e-02 0.000 1.103040 0.330180 -0.751070 1.0 1.0 1.482259 168.933 2.800 0.200 4.389696 14528.1218
C W C 1.0 2.081300e-04 0.000 330.000000 3.500000 -1.000000 1.0 0.0 0.000000 0.000 1.850 0.150 0.000000 0.0000
C C W 1.0 7.285500e-02 0.000 1.103040 0.330180 -0.751070 1.0 0.0 0.000000 0.000 2.800 0.200 0.000000 0.0000
W C W 1.0 1.882270e-03 0.459 2.149690 0.171260 0.277800 1.0 0.0 0.000000 0.000 3.500 0.300 0.000000 0.0000
1 Like
You can always upload files to dropbox, google drive, or one-drive or equivalent and post a link.
When putting files inline, you should quote them with “```” so the forum does not try to typeset them. Please see my edit to your post.
It seems to me the most likely explanation is that your initial crystal structure is incorrect. Isn’t WC a hexagonal system? The Tungsten Carbide (B_h) Structure
When I change the custom lattice according to the settings from that website I get the following and that is stable at 300K.
You are right, my previous understanding of the structure of WC was wrong. I read the lattice command again, combined with the website you provided, and modified the lattice command as follows, hope it’s right:
variable a equal (1/2)*sqrt(3)
variable b equal 2/3
variable c equal 1/3
#MODEL
lattice custom 2.926 &
a1 0.5 -${a} 0 &
a2 0.5 ${a} 0 &
a3 0 0 1 &
basis 0.0 0.0 0.0 &
basis ${b} ${c} 0.5
At 300K, the W atoms of the model’s boundaries shift a little, but overall the model is stable:
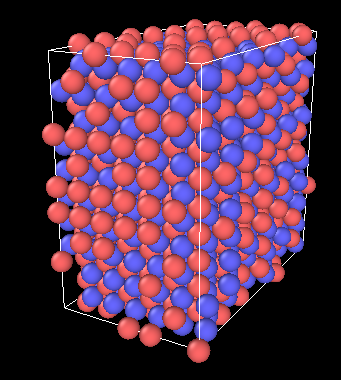
Thank you for your time and effort in helping me find the error 
I think you can further improve your simulation by:
- changing your primitive cell in the custom lattice to apply the a/c ratio of 0.98
- doing a minimization with fix box/relax before the MD
- use anisotropic cell change instead of enforcing isotropic change (which is most meaningful with liquids)
Your image still shows atoms that move to places that are not as close to the original lattice due to the volume constraints.
Hi, I’m sorry for taking so long to reply to you, I’ve been busy writing my paper these days 
Thanks to your suggestion, I changed the a/c ratio and added the fix box/relax command as follows.
units metal
atom_style atomic
boundary p p p
neighbor 0.3 bin
variable a equal (1/2)*sqrt(3)
variable b equal 2/3
variable c equal 1/3
variable d equal 2.853/2.928 # a=2.928, c=2.853
#MODEL
lattice custom 2.926 &
a1 0.5 -${a} 0 &
a2 0.5 ${a} 0 &
a3 0 0 ${d} &
basis 0.0 0.0 0.0 &
basis ${b} ${c} 0.5
region box block 0 6 0 6 0 10 units lattice
create_box 2 box
create_atoms 2 box basis 1 1 basis 2 2
#MASS
mass 1 183.84 #W
mass 2 12.01 #C
#POTENTIAL
pair_style tersoff
pair_coeff * * WC.tersoff W C
#THERMO
thermo 1000
thermo_style custom step temp pe vol press
#minimize
fix relax all box/relax iso 0.0 vmax 0.001
minimize 1.0e-12 1.0e-12 10000 10000
unfix relax
#300K, relax
velocity all create 300 66666 rot yes dist gaussian
fix 1 all npt temp 300 300 0.1 iso 0 0 1
dump 1 all custom 1000 WC_relax.lammpstrj id type x y z vx vy vz
timestep 0.001
run 10000
The figure below shows the result of relaxation at 300K, some atoms are still displaced (I don’t quite understand why, maybe I wrote the wrong command?)
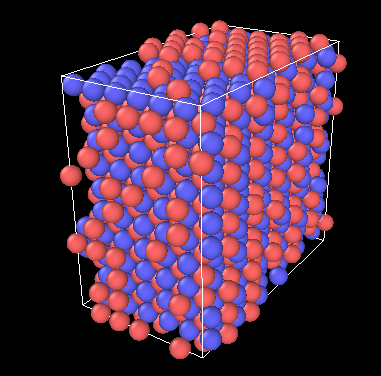
Also, I don’t quite understand how to implement the “use anisotropic cell change instead of enforcing isotropic change” you mentioned. I want to simulate the WC in a high temperature environment and hope that it will at least partially melt. It would be great if you could provide me with some further explanation
This is due to periodic boundary conditions and the fact that you have placed your lattice so that atoms are created exactly on the boundaries. You can avoid this by either shifting the box boundaries or the origin of the lattice.
This means you have not read the documentation for fix box/relax and fix npt with sufficient care and attention to detail.
Simulating melting in atomic scale MD simulations is tricky business since melting is an activated process that depends on fluctuations and thus is subject to finite size effects. This typically results in a very large hysteresis. It is therefore usually a good idea to stay away from simulations in the neighborhood of melting points. For studies of the melting point directly, it is usually a good idea to consider coexistence studies.