Hi everyone,
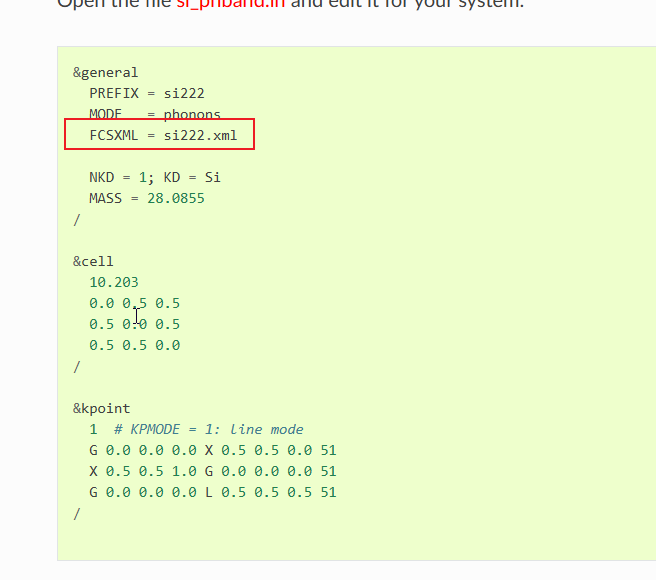
After checking the ALAMODE official website, I found that calculating the phonon spectrum requires a specific format harmonic force constant file.
However, during the process of calling Alamode with GULP, the output is a third-order force constant file.
I would like to ask how to use GULP to generate the second-order force constant file in a format readable by Alamode, or whether it’s possible to use the third-order force constant file to plot the phonon spectrum as well. Here are my attempts:
You may need to ask Alamode for help with their file formats. However, as the thermal conductivity calculation with GULP-Alamode generates phonons there should be a file from Alamode with force constants there. If there are differences in the phonons for SiC then this could be because the force field input isn’t quite the same as the reference you are comparing against. GULP computes the second derivatives analytically and doesn’t need a supercell, whereas other combinations of codes (e.g. LAMMPS) that only have 1st derivatives will require large enough supercells to convergence the force constant matrix in the supercell (though for Tersoff, for example, this would be quite short ranged).
Hi,Julian,
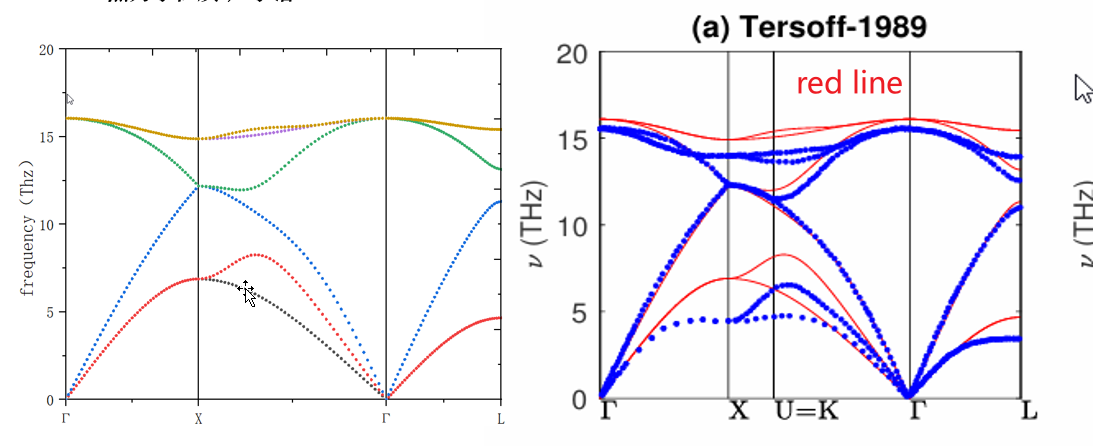
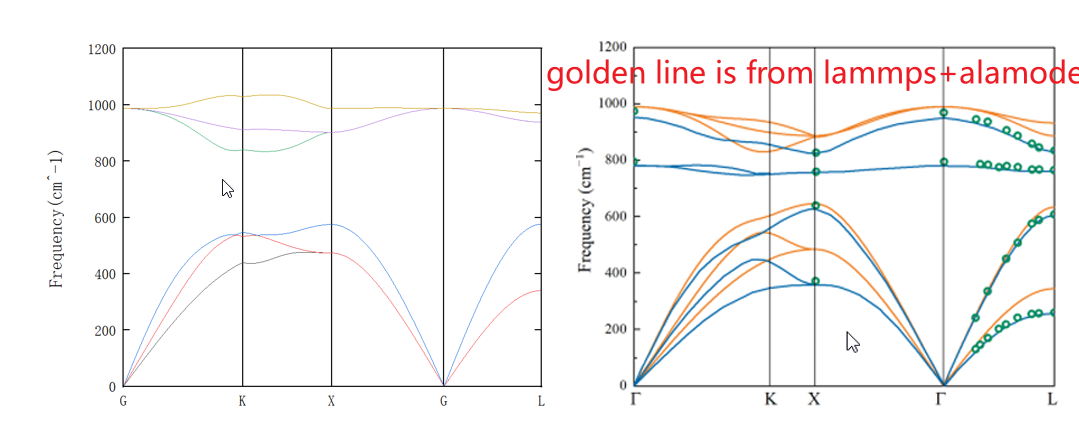
Both Si and SiC use the Tersoff potential, and the compared phonon dispersion are also based on Tersoff. So I am confused about why Si fits well while SiC does not.
If the potential model input is different for SiC (regardless of whether it’s Tersoff) then the results can be different. A guess might be that the cross-terms for Si-C are being handled differently, such the end result is not the same for both codes. You can see if this might be case by running pure C as well.