Hi all,
Question 1: I would like to know if there is a way to generate specific terminations for a metal oxide crystal structure like SiO2 - (0,0,1).
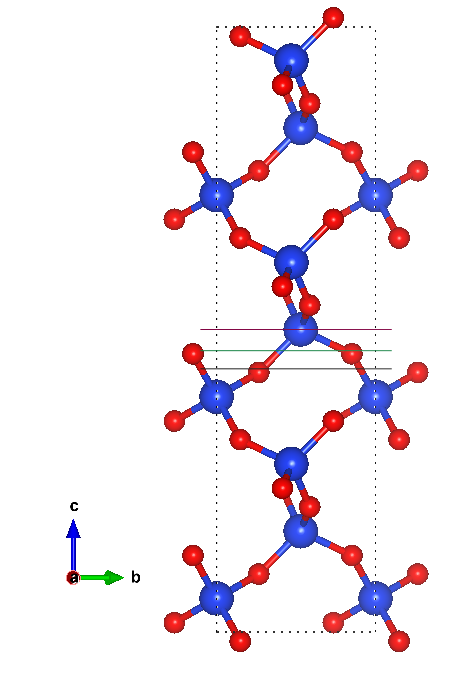
Below is an example of the terminations that I would like to study. I tried both methods SlabGenerator and generate_all_slabs. When I turned symmetrize = True, I got 0. I got 2 slabs when I turned symmetrize off but it didnt cur the slab at the terminations of interested. I tried to change the min_slab_size, it only increased the number of layers, still provide the same termination.
Question 2: I would like to know if there is a method in pymatgen to generate slab with different number of layers. For example, 6,9,12,15,18,19,20…layers of the SiO2 slabs. When I increased or decreased the min_slab_size, I either got double, triple, or same initial layer of the slab in z-direction.
Thank you for everyone’s time!
Hi @Ngan_Huynh,
This is interesting. I tried reproducing your issue with the conventional cell of SiO2 (mp-546794) when producing slabs with (0,0,1) orientation. It turns out that I also get only 1 slab when using the function get_slabs(). I looked into the source code and it seems that _calculate_possible_shifts returns 8 possible terminations but StructureMatcher classifies all 8 of those to be equivalent. I think this to be wrong because with two atom types there should at least be 2 unique terminations (but actually there should be even more than that). Is this potentially a bug of StructureMatcher @Richard_Tran?
As for your second question. You can set the flag in_unit_planes=False in SlabGenerator and then the min_slab_size refers to Angstroms rather than number of unit cells. However, I believe, SlabGenerator will never return a slab that is less than 1 unit cell in width. In that case, you’d need to manually delete layers.
Best,
Peter
Hi,
Thank you for your time to reproduce the issue and the suggestion for question 2. I will give it a try. I used to cut the surface by hand and ran structure optimization to compare the energy and get the lowest one. My college recommended me to give pymatgen a try and I love how quick it was for me to generate many slab structures. However, the cutting at specific terminations is still an issue for me since I need to prove to my advisor that the one from pymatgen was the most stable one.
Thank you again for your help!
Best,
Ngan Huynh
Do you have a cif file or mpid for the structure? I don’t think its the same type of SiO2 as mp-546794. In any case, I played around with the mp-546794 working under the assumption that there are three possible terminations as you suggested in the image:
- Si with no O coordinated on top
- Si with 1 O coordinated on top
- Si with 2 O coordinated on top.
Using an ftol=0.01 (this allows for a finer search of shift values), I obtained two slabs:
- A slab with an Si-O on both surfaces
- A slab an Si-O2 termination on one surface and an Si termination on the other.
This is where your three terminations are. I’ve also double check all the possible slabs generated with all possible shift values regardless of StructureMatcher and confirmed that these 2 slab types are the only unique ones to appear so I don’t think StructureMatcher is the issue here.
Having said that, here is the problem, the way SlabGenerator creates slabs by default is by first create the vacuum and slab layer and then afterwards, shifting whatever atoms on one surface above the shift value to the other surface.
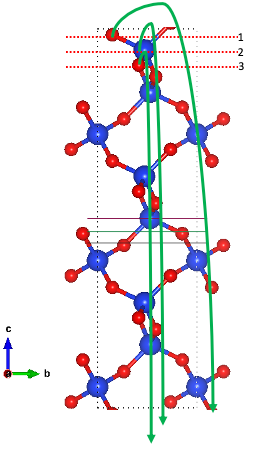
Have a look at at the image above. For convenience, I moved the termination lines to the top. Now if I move (1) to the other side, I will end up with two terminations (Si with no O coordinated on top
and Si with 2 O coordinated on top). In other words, using this method of slab generation will not be able to produce a slab of these two terminations where both surfaces are the same. You will run into the same problem when shifted (2). If I shift (3), this will create a slab where both surfaces have a Si atom with an O atom on top.
If you want to get (1) and (2) which are unique terminations on their own, you have two options:
- Perform a Tasker type reconstruction (this requires you expand the surface area of your slab, something like slab.make_supercell([2,2,1]) and then use the get_tasker2_slabs() Slab method). However this will generally create a completely different termination from the one you want anyways.
- You remove an atom on the other side to get the same surface. Note that this leaves you with a non-stoichiometric solution. If you plan on getting the surface energy with this termination, you’ll have to compensate the missing species with the chemical potential.
Usually, option 2 is taken care of if you set symmetrize=True which ignores stoichiometry and just removes sites until it obtains a slab with Laue symmetry. However Laue symmetry is only a sufficient but not necessary condition for checking if your two surfaces are equal. I imagine that when using symmetrize=True, it is generating those two other terminations however its ignoring them due to the lack of Laue symmetry. This is just another contingency of surface construction I need to fix in a later release, but for now if you want to get these nonstoichiometric terminations, I suggest manually remove the atom at the surface to get the termination you want. You can do this by taking the unequivalent slab and removing the lone Si on one side to get a 2O terminated slab or remove the two O atoms on the other side to get a lone Si terminated slab.
Hi Richard,
Thank you for the explanation.
I tried to read the source code to understand how SlabGenerator generate the slab. I will play around with the structure to see if I can reproduce 2 slabs.
The mpid i used was mp-6930 - I think this is alpha-SiO2. I looked up the phase diagram of SiO2 and found that SiO2 will have alpha phase for the temperature range I am studying.
Now you pointed out the mpid, may be the structure I used wasn’t good that’s why it could not relax and give me final energy during structure relaxation in VASP.
Thank you again for your time!
Ngan Huynh
I don’t know if either mpid correspond to the structure you were looking for, I was just pointing out a disclaimer that the example structure I was using (mp-546794) was not the same structure as yours, but nonetheless, I think the example should still hold for both cases. Good luck.
@Richard_Tran
Thanks for the detailed explanation! That does make sense. For some reason it’s still giving me only one slab for mp-546794 (SlabGenerator(structure, [0,0,1], min_slab_size=2, min_vacuum_size=5).get_slabs(ftol=0.01)) but it’s probably fine.
I’m more curious about the other comments you mentioned: Do you have advice regarding the procedure on how to pick the chemical potential for non-stoichiometric slabs? Or maybe a paper you could refer us to? (I have read your paper and the Sun & Ceder surface creation paper).
Again, thanks for your insights!
-Peter
I usually set my slab size to at least 10-15Å thick, so maybe that’s why it was able to find two terminations. In any case, Rogal gives a pretty good explanation on calculating the chemical potential limit for non-stoichiometric surfaces here:
Essentially your range is determined by the enthalpy of reaction for SiO2. You can get the chemical potential range pretty easily using the PhaseDiagram class method .get_chempot_range_map().
Great, thanks so much for the reading material, @Richard_Tran! Highly appreciate it!
Hi all,
Thank you for the suggestions!
I tried and got 2 slabs for mp-546794.
I used
slabgen = SlabGenerator(sio2_struct,miller_index=[0,0,1],min_slab_size=8,min_vacuum_size=15,center_slab= True)
all_slabs = slabgen.get_slabs(ftol = 0.01)
Another quick question relate to structure on Material Project, I would like to use alpha-Quartz and beta-Quartz for my optimization. Do you happen to know which material id to use? There are many on the site for SiO2 and I am not sure which one is correct.
Thank you again for your time!
Ok, I think I found the difference. I used the conventional cell for mp-546794 and that gives only 1 slab, if I use the primitive cell, then it also gives me 2 slabs. Interesting…
I am pretty sure that mp-6922 is beta-quartz because it matches the spacegroup (nr. 180) and it also mentions that it is beta quartz in the comments at the bottom of the page. For alpha-quartz there are two structures that match the spacegroup (nr. 154): mp-640917 and mp-6930. It’s probably mp-6930 because the lattice parameters are closer to the experimental values of alpha-quartz.