Hi there, I am trying to figure out how lammps process the p p p boundary condition.
Suppose that I try to model the bulk behavior of graphite through a 2-layer graphene with p p p b.c…
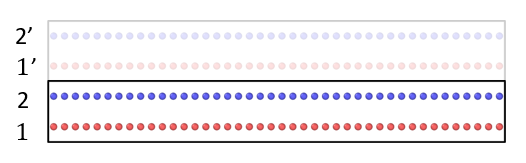
Atoms within the two layers are labeled by type 1 and 2 (shown in the picture below), respectively. The intralayer interation is modeled by rebo and interlayer by LJ, as
To process the ppp b.c., there is an imaginary box labeled by (‘). Would layer 1 and layer 1’ be different to lammps? In other words, would there be interaction between layer 1 and 1’, since only pair_coeff between layer 1 and layer 2 are specified?
Would there be more imaginary boxes as needed, if I increase the cutoff distance of LJ, say, to 20? Under such circumstance, the cutoff distance would cross more than 2 boxes.
Ghost atoms have the same atom type than their original, so there will be interactions computed.
The largest cutoff determines also the ghost atom cutoff and that may be more than half the box dimension since LAMMPS is not subject to minimum image conventions. It uses real copies of the ghost atoms and updates their positions during the simulation and does not need to find the closes image during the force computation. The choice is already made during the neighbor list construction.
This is just what confuse me.
If ‘ghost atoms have the same atom type than their original’, then atoms in layer 1 and layer 1’ have the same atom type 1. This would lead to no interactions between layer 1 and layer 1’, because there is no LJ pair_coeff specified between type 1 and type 1. Of course, the interactions between layer 1’ and layer 2 will be computed, since they have different atom type. Hence, to successfully model the bulk behavior, the box should contain enough layers such that the total thickness of all layers is not smaller than the cutoff distance.
Is this understanding correct?
I have this confusion, because I am trying to understand the layer-number-dependent total potential encountered in my simulation. Simply take the system above as an example, during shearing the box parallel to the graphene surface by ‘fix deform’, the total potential energy is calculated and then averaged by the number of layers n contained by the box, which is denoted by En. The above example containing 2 layers corresponds to E2. I find that En decreases sharply at first and soon becomes steady, as n increases from 2.
It appears that the above mentioned understanding about ppp b.c. processing might explain this odd phenomenon.
If you want a different behavior, you need to set up a larger system with more layers. Then each layer has a different atom type and you can apply Lennard-Jones to the inter-layer interactions.