Hello all,
I want to simulate an indentation (Carbon on Nickel). I divided each block into three atoms types.
1*3: Ni (eam/alloy)
4*6: C (rebo)
but I have some problem using pair_style hybrid.I’m using the following lines to do so:
(the file that I’m using for Ni works correctly, and please don’t mention its reliability.)
pair_style hybrid eam/alloy rebo lj/cut 6
pair_coeff * * eam/alloy Ni.lammps.eam.alloy Ni Ni Ni NULL NULL NULL
pair_coeff * * rebo CH.airebo NULL NULL NULL C C C
pair_coeff 3 4 lj/cut 0.1 2.138
Later the code calculates the potential energy per atom and dump it:
compute pea all pe/atom
fix OUT all ave/atom 1 {ouTme} {ouTme} c_pea
dump 1 all custom ${ouTme} dump.lammpstrj id type xs ys zs f_OUT
The output (attached to this email) shows that there is a problem either with the method I’m using the potential energy, or its extraction, or both.
Is there any obvious problem in the code, I’m missing?
Thanks in advance,
Soheil
PS_1. The temperature is held at 300 K. and I’m using LAMMPS 64-bit 2014.05.19.
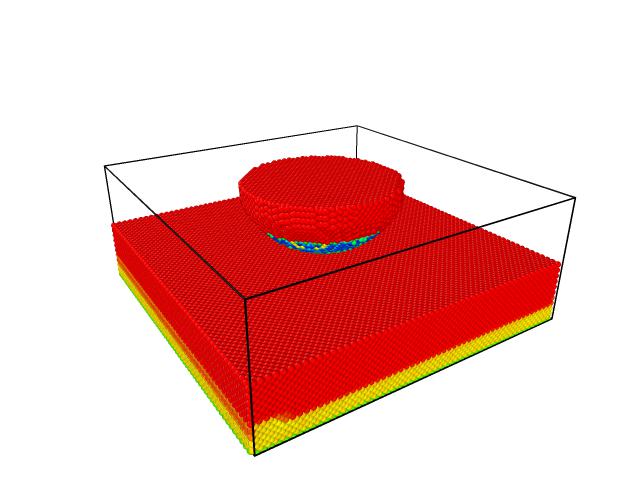
PS_2. The attached snapshot shows the potential energy of the system in the range of (red: 0 eV, blue:-7.7eV)