I constructed a starch-water system in Materials Studio and convert the MS file to LAMMPS data file by the msi2lmp tool. After MD simulation in LAMMPS, I performed the hydrogen bond analysis in VMD. But I failed to see any hydrogen bonds in the LAMMPS trajectory file, even if I increased the Donor-Acceptor distance to 100 angstroms and the Angle cutoff to 100 degrees.
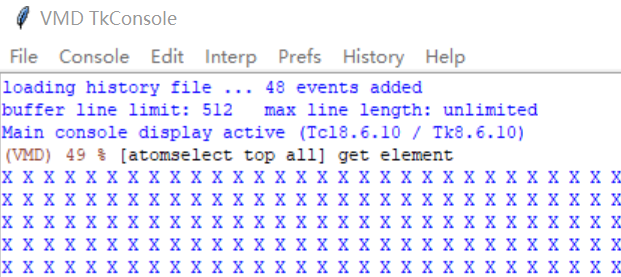
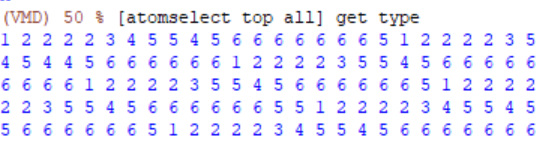
I guess if it’s because VMD failed to identify the element type of the atoms from the LAMMPS trajectory file correctly (type 1,2,3 are carbon, type 4,5,8 are oxygen, type 6,7,9 are hydrogen).
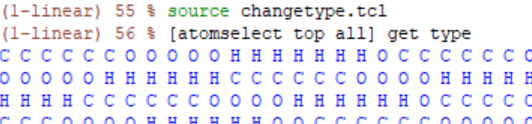
So I modified the element, type and name attributes of the atoms in VMD with “set” command.
But I still failed to see any hydrogen bonds using the hydrogen bond analysis tool in VMD.
From your information we can’t tell if your simulation failed (wrong force field / bad initial configuration / simulation too short) or if the simulation was successful but the hydrogen bond analysis tool failed (for example it couldn’t read atom elements correctly) – or if there is a bug in either LAMMPS or VMD, but these are far less likely (though very much possible).
You should create a movie of your simulation trajectory and watch it to see at least if there are any obvious problems – and if you can identify visually some H-bonds that should be detected then you know you aren’t invoking the H-bond analysis tool successfully.
The following information might be helpful to us:
your LAMMPS version
your input script
simulation features (did energies suddenly change? Was temperature well-controlled?)
a system snapshot
But you’re likely to get more help if you can pinpoint the exact step where something is going differently from what you would expect.
This is really a question for the VMD mailing list and not a LAMMPS forum.
For most of the advanced features in VMD you have to understand that it was primarily written for use with NAMD which was primarily written for use with the CHARMM force field and systems where the atom naming follows the rules of the protein database (PDB). So there are plenty of cases when heuristics assume that inputs follow the conventions of those force field and geometry file formats. This applies to the hydrogen bond tool as well.
It uses the atomselect macro “hydrogen” as atom selection with the text name "[0-9]?H.*", so your changing of the “type” property does not help with that, you need to change “name”.