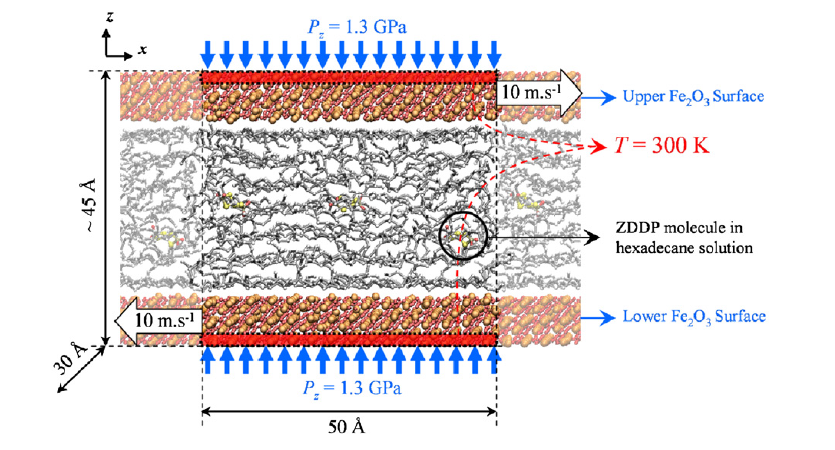
This looks like a very complex simulation yet your question indicates that you very little experience with setting up simulations. I therefore suggest you discuss with your adviser or tutor how you will build your skills in stages toward the goal of setting up a such a simulation. If you start with this kind of system right away, you will never know where issues are coming from, since there are too many possible sources and the number of possible errors multiplies with the degree of complexity.
Please also note that for such complex system, creation of the geometry is only a (minor) part of the total problem. You also need to find suitable force fields and force field parameters, figure out how to assign atom types, derive a topology, assign bond, angle, dihedral etc. types, handle how to apply external pressure and more.
As a general approach I would suggestion you follow these stages:
- set up a bulk hexadecane molecular system and reproduce published data of its properties (from simulation!)
- set up a bulk haxadecane ZDDP mixture and reproduce published data, if available
- set up a bulk Fe2O3 system and reproduce published data (from simulations)
- set up a single Fe2O3 slab system (oxides tend to require force fields with charge equilibration which are difficult to control, specifically for systems with surfaces). observe how to best “cut” the slabs to minimize unphysical dipoles across the slab.
- If you need to use charge equilibration for Fe2O3 (which is likely), you need to determine how large a region of atoms with fixed positions and bulk charges you need to emulate bulk and where you can cut
- set up a single Fe2O3 slab with hexadecane and sort out how you need to properly model the interface (there is not likely going to be a single
- study how to set up a simple dual slab system (say copper with liquid argon in between) and how to apply pressure. As alternative consider using periodic boundaries in z-direction and fix deform or fix npt to compress in z-direction
- set up a dual slab system as shown in your figure or alternately consider setting up a periodic
Setting up the individual atomic/molecular components can be done with LAMMPS itself or a variety of external tools. There are discussions about benefits and simulation protocols for such multi-slab z-periodic systems in the archives. So a look through those would not hurt.
Bottom line, this is not a simulation that can be done with following simple “do this, not that” instructions. Instead, it is necessary to validate each step and each component and there are many ways to make mistakes. Not all mistakes will lead to errors, some will just cause unphysical results.
@akohlmey, Thank you for your kind reply. If i want to create whole crystal structure of the same model than which software can I used and the 3D crystal structure later convert to data file for LAMMPS.
There is no single “do this, not that” style of approach for this. You may be able to build the entire structure with LAMMPS, or use external pre-processing and system building tools (several are liested on the LAMMPS homepage).
But it will not likely work in the way that you have a tool that will create the entire structure and you can just “convert” it and run your simulations. The system you have shown is quite complex and thus one have to approach it in stages, you need to develop a “workflow strategy” to approach an equilibrated structure in gradually. No tool is able to build such a complex structure in an equilibrated state directly and without pre-equilibration, the simulation will likely not succeed. At all these steps it is crucially important not only to worry about the geometry, but also about the force field(s) in use and the implication of those choices to building topologies and assigning atom, bond, angle, dihedral types and so on.
Yes thank you for your valuable suggestion. @akohlmey.