Dear @magnusrahm
The xyz file format works!
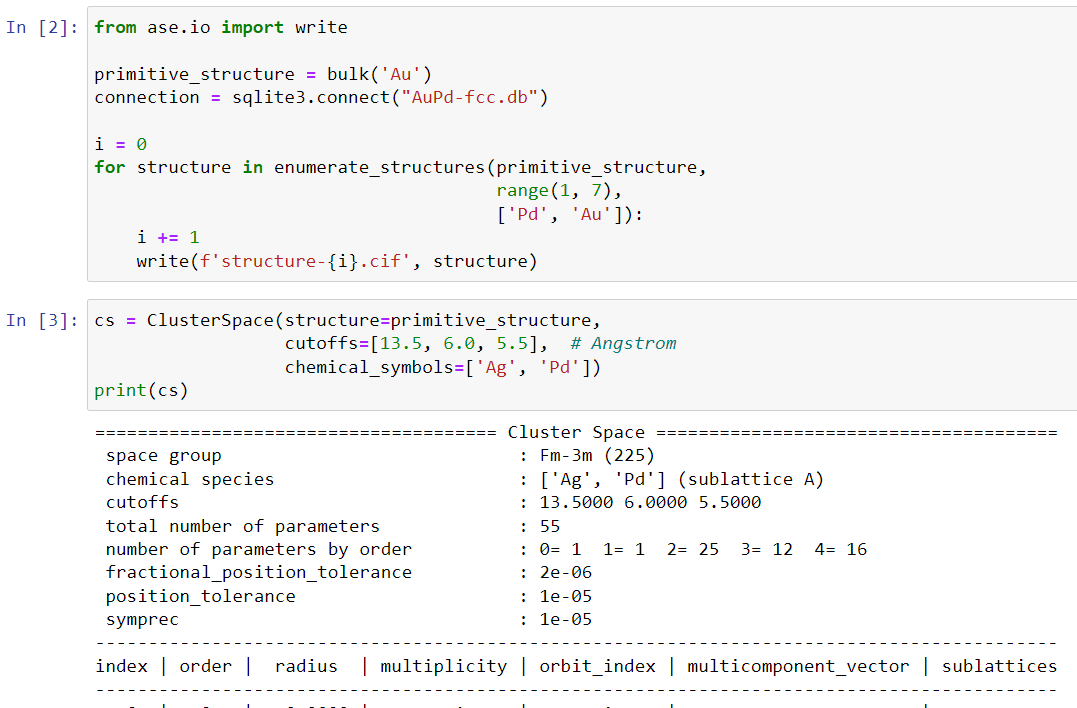
from ase.io import write
primitive_structure = bulk('Ag')
connection = sqlite3.connect("AgPd-fcc.db")
i = 0
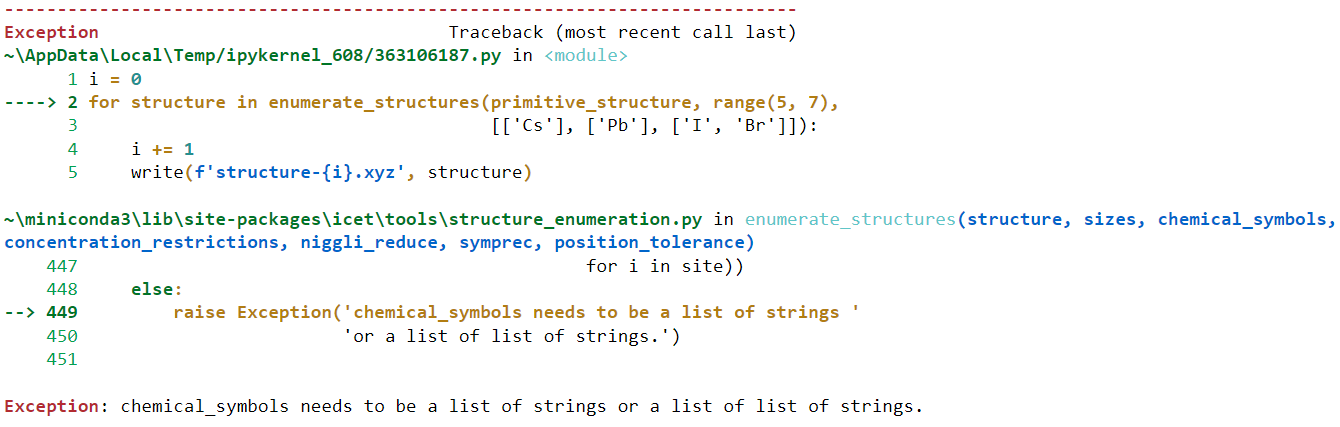
for structure in enumerate_structures(primitive_structure,
range(1, 7),
['Pd', 'Ag']):
i += 1
write(f'structure-{i}.xyz', structure)
cs = ClusterSpace(structure=primitive_structure,
cutoffs=[13.5, 6.0, 5.5], # Angstrom
chemical_symbols=['Ag', 'Pd'])
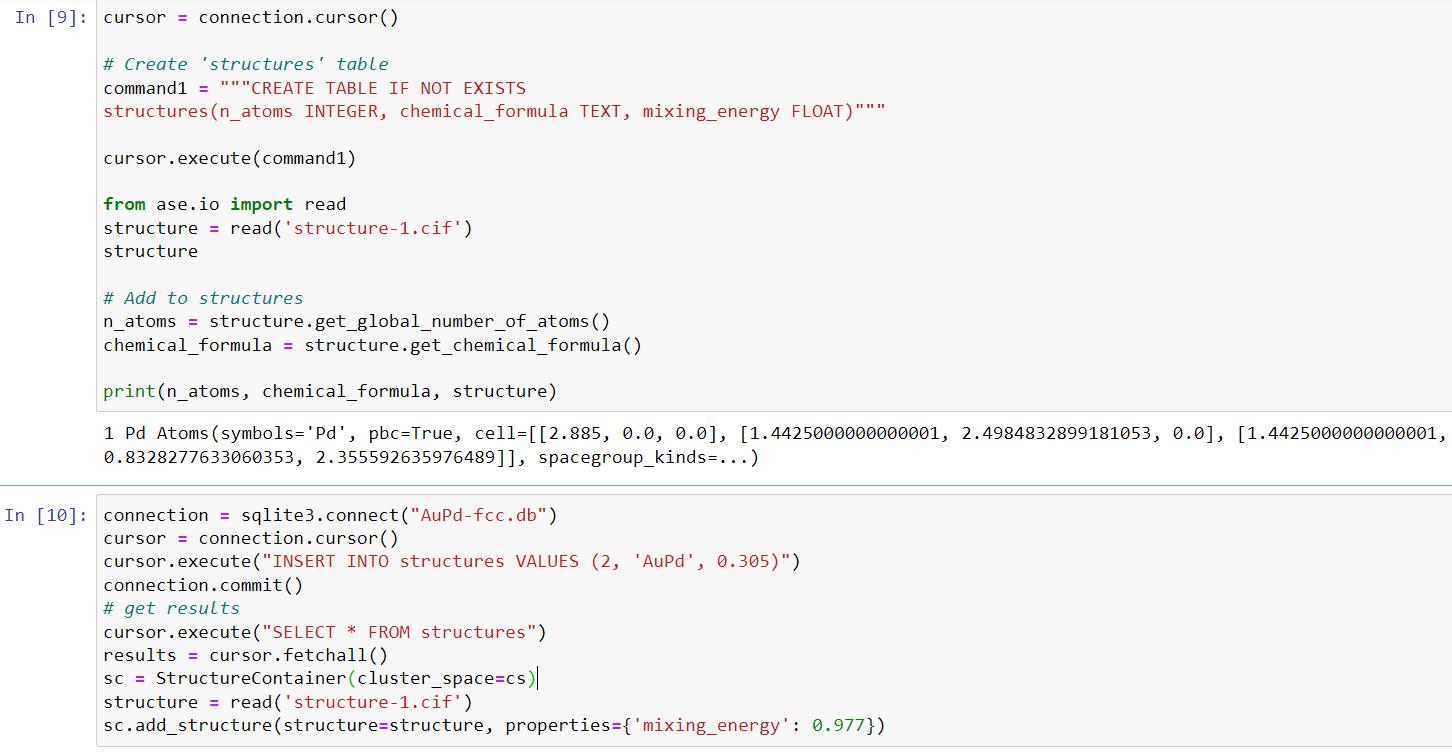
connection = sqlite3.connect("AgPd-fcc.db")
cursor = connection.cursor()
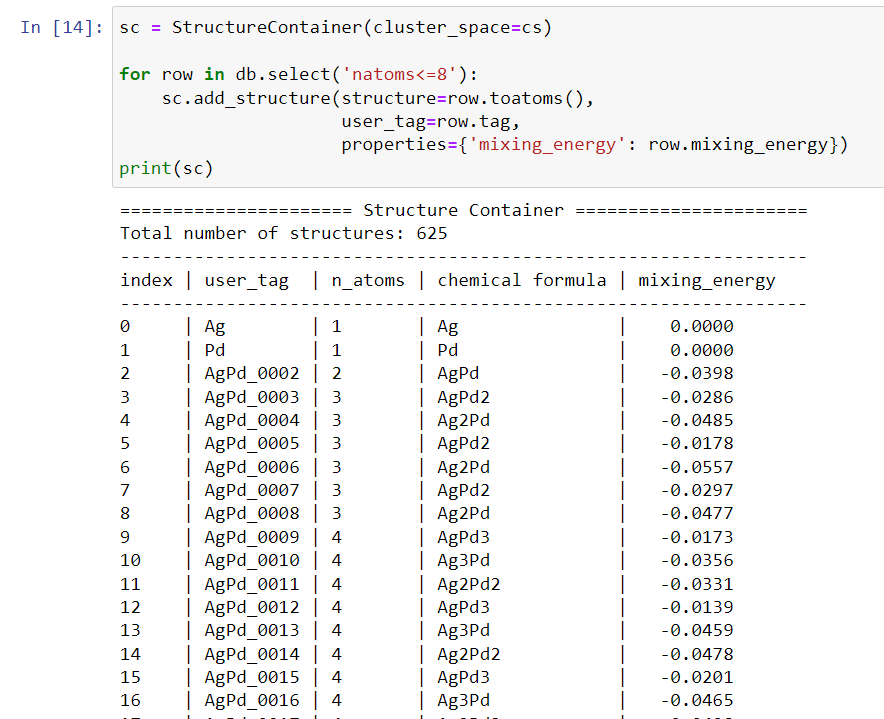
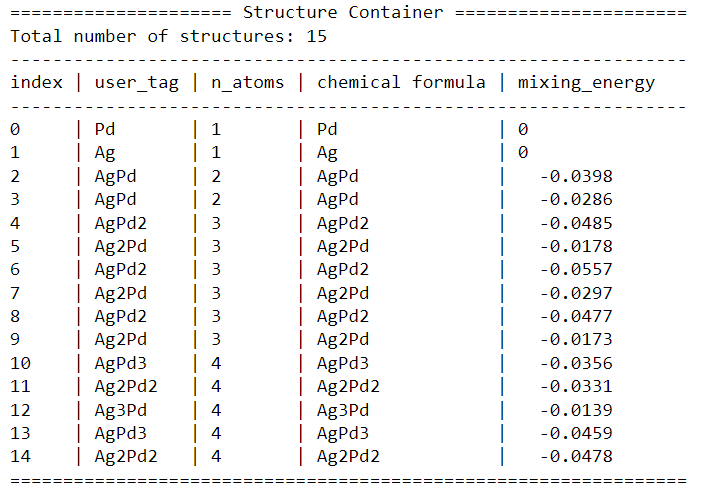
sc = StructureContainer(cluster_space=cs)
mixing_energy = [0, 0, -0.0398, -0.0286, -0.0485, -0.0178, -0.0557, -0.0297,
-0.0477, -0.0173, -0.0356, -0.0331, -0.0139, -0.0459, -0.0478]
for i in range(len(mixing_energy)):
structure = read('structure-{}.xyz'.format(i+1))
sc.add_structure(structure=structure, user_tag=structure.get_chemical_formula(),
properties={'mixing_energy': mixing_energy[i]})
print(sc)
opt = CrossValidationEstimator(fit_data=sc.get_fit_data(key='mixing_energy'),
fit_method='lasso')
opt.validate()
opt.train()
ce = ClusterExpansion(cluster_space=cs, parameters=opt.parameters, metadata=opt.summary)
ce.write('mixing_energy.ce')
ce = ClusterExpansion.read('mixing_energy.ce')
data = {'concentration': [], 'reference_energy': [], 'predicted_energy': []}
db = connect('AgPd-fcc.db')
for row in db.select():
data['concentration'].append(row.concentration)
# the factor of 1e3 serves to convert from eV/atom to meV/atom
data['reference_energy'].append(1e3 * row.mixing_energy)
data['predicted_energy'].append(1e3 * ce.predict(row.toatoms()))
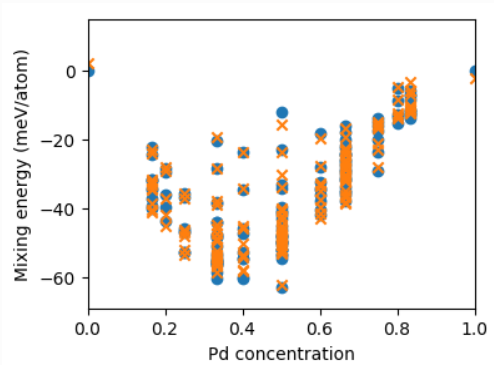
fig, ax = plt.subplots(figsize=(5, 4))
ax.set_xlabel(r'Pd concentration')
ax.set_ylabel(r'Mixing energy (meV/atom)')
ax.set_xlim([0, 1])
ax.set_ylim([-69, 15])
ax.scatter(data['concentration'], data['reference_energy'],
marker='o', label='reference')
ax.scatter(data['concentration'], data['predicted_energy'],
marker='x', label='CE prediction')
plt.legend()
plt.show();
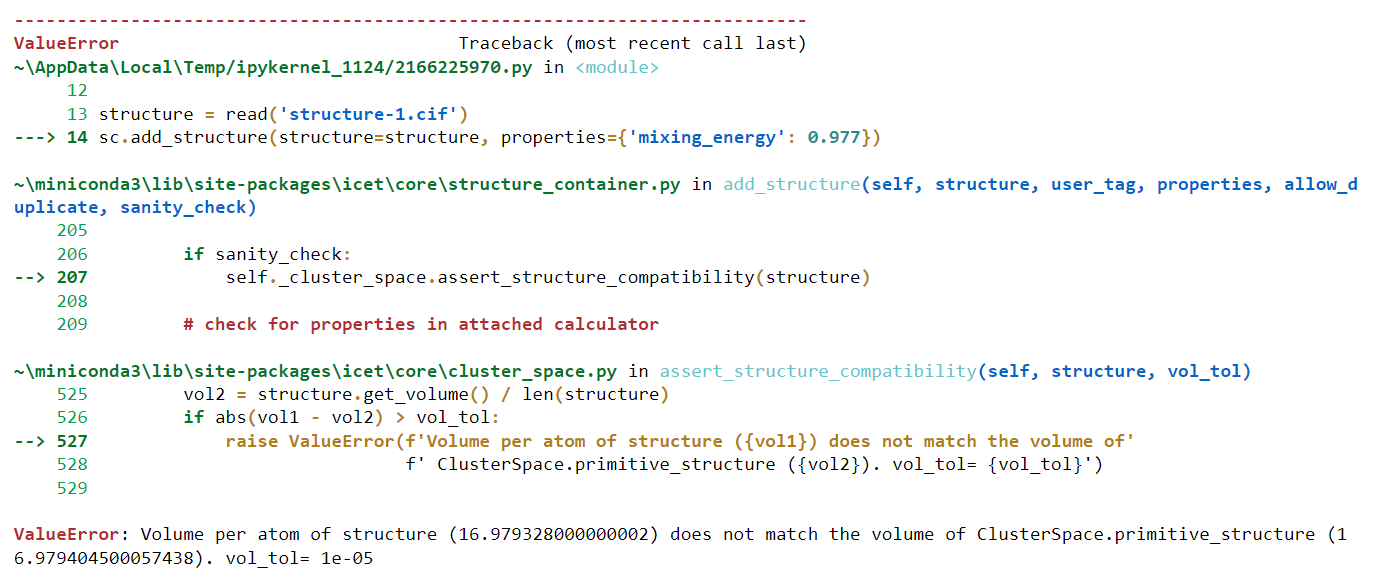
I’m really confused about the last part of the code. How do I get the reference_energy?